BioAccord LC-MS システムおよび INTACT Mass waters_connect アプリケーションを使用した RNA の CQA 分析
研究目的のみに使用してください。診断用には使用できません。
要約
このアプリケーションノートでは、リボ核酸(RNA)ベースの医薬品の重要品質特性(CQA)を分析するための新しいインフォマティクス機能を紹介します。BioAccord™ LC-MS システムを使用し、waters_connect™ インフォマティクスプラットホームの制御下で、イオン対逆相(IP-RP)液体クロマトグラフィー質量分析(LC-MS)データを取り込みました。mRNA Cleaver マイクロアプリ(in silico フラグメント生成用)、INTACT Mass アプリ(RNA 消化産物の検出および割り当て用)、Coverage Viewer マイクロアプリ(消化データのカバレッジの視覚化用)などのアプリケーション固有のソフトウェアが開発されました。このワークフローは、新世代の医薬品およびワクチンに一般的なシングルガイド RNA(sgRNA)およびメッセンジャー RNA(mRNA)の両方の CQA について実証されています。
アプリケーションのメリット
- データ解析用の新しいバージョンの INTACT Mass アプリおよび消化物フラグメントの in silico 生成とキュレーションのための追加のマイクロアプリツールを使用した、RNA 消化フラグメントマッピング、核酸消化産物の質量確認のためのデータ分析ワークフロー
- mRNA の 5' キャッピング効率および 3' ポリ(A)テールの不均一性を評価するための INTACT Mass アプリを使用する追加のワークフロー
はじめに
ここ 10 年で、RNA ベースの医薬品およびワクチンの開発が急増しました1-3。 新型コロナウイルスに対する最近の 2 種類の mRNA ベースのワクチンの開発および承認により、RNA ベースの医薬品がバイオ医薬品業界の最前線に押し出されました4-6。 そのため、RNA ベースの医薬品の製造プロセスを適切に管理し続けるために、CQA をモニターするための分析法の開発の優先度が高くなっています。核酸医薬品の CQA には、配列確認、5' キャッピングの効率(mRNA)および構造、3' ポリ(A)末端の不均一性(mRNA)の分析、修飾ヌクレオチドの位置決定、活性 RNA 生成物の純度評価などが含まれています。
LC-MS システムは、RNA ベースの医薬品の CQA を特性解析する際の有効なツールとして確立されています 7-10。ただし、データ解析が共通のボトルネックであり、これに対処する必要があります。このアプリケーションノートでは、waters_connect インフォマティクスプラットホームで開発されたツール(特に INTACT Mass、mRNA Cleaver、Coverage Viewer の各アプリケーション)を使用した RNA の CQA 分析のためのデータ解析ワークフローについて説明します。
実験方法
試薬およびサンプル前処理
今回報告する実験条件は、sgRNA の消化および配列マッピング実験用のものです。このアプリケーションノート内の mRNA の CQA データ生成に適した実験の詳細およびサンプル情報は、関連するウォーターズのアプリケーションノート(mRNA マッピングデータ(Gaye ら)、3' Poly(A)テールのデータ(Doneanu ら)、5' キャップのデータ(Ngyuen ら))に記載されています13,14,18。
データ解析ワークフローの例:
N,N-ジイソプロピルエチルアミン(DIPEA、純度 99.5%、カタログ番号 387649-100ML)、1,1,1,3,3,3-ヘキサフルオロ-2-プロパノール(HFIP、純度 99%、カタログ番号 105228-100G)、エタノール(HPLC グレード、カタログ番号 459828-2L)、重炭酸アンモニウム(LiChropur LC-MS Supelco 試薬、カタログ番号 5330050050)は Millipore Sigma(ミズーリ州セントルイス)から購入しました。アセトニトリル(LC-MS グレード、カタログ番号 34881-1L)およびメタノール(LC-MS グレード、カタログ番号 34966-1L)は、Honeywell(ノースカロライナ州シャーロット)から入手しました。HPLC グレードのタイプ I 脱イオン水(DI)は、Milli-Q システム(Millipore、マサチューセッツ州ベドフォード)を使用して精製しました。移動相は毎日新しく調製しました。sgRNA 消化用のヌクレアーゼフリー超高純度水(カタログ番号 J71786.AE)は、Thermo Fisher Scientific(マサチューセッツ州ウォルサム)から購入しました。
100 mer の sgRNA は IDT(Integrated DNA Technologies、アイオワ州コーラルビル)から購入し、ヌクレアーゼフリー水 100 µL に再溶解しました。sgRNA には、配列の末端の位置 1、2、3 および 97、98、99 に、6 つの安定性を高める修飾ヌクレオチドが含まれています(図 1A)。含まれている修飾ヌクレオチドは 2'O-メチルグアノシン、2'O-メチルウリジン、2'O-メチルアデノシンで、各修飾にチオリン酸基が含まれています(図 1B)。RNase T1 リボヌクレアーゼは Worthington Biochemical Corporation(ニュージャージー州レイクウッド)から購入しました。sgRNA の消化では、5 µL の再溶解した sgRNA を消化バッファー(100 mM 重炭酸アンモニウム)1 µL、ヌクレアーゼフリー水 43 µL、および 1 mg/mL RNase T1 溶液(ヌクレアーゼフリー水中で調製)1 µL と混合しました。この消化混合液を QuanRecovery™ MaxPeak™ 300 µL バイアル中で調製し、37 ℃ で 15 分間インキュベートした後、直ちに LC-MS で分析しました。
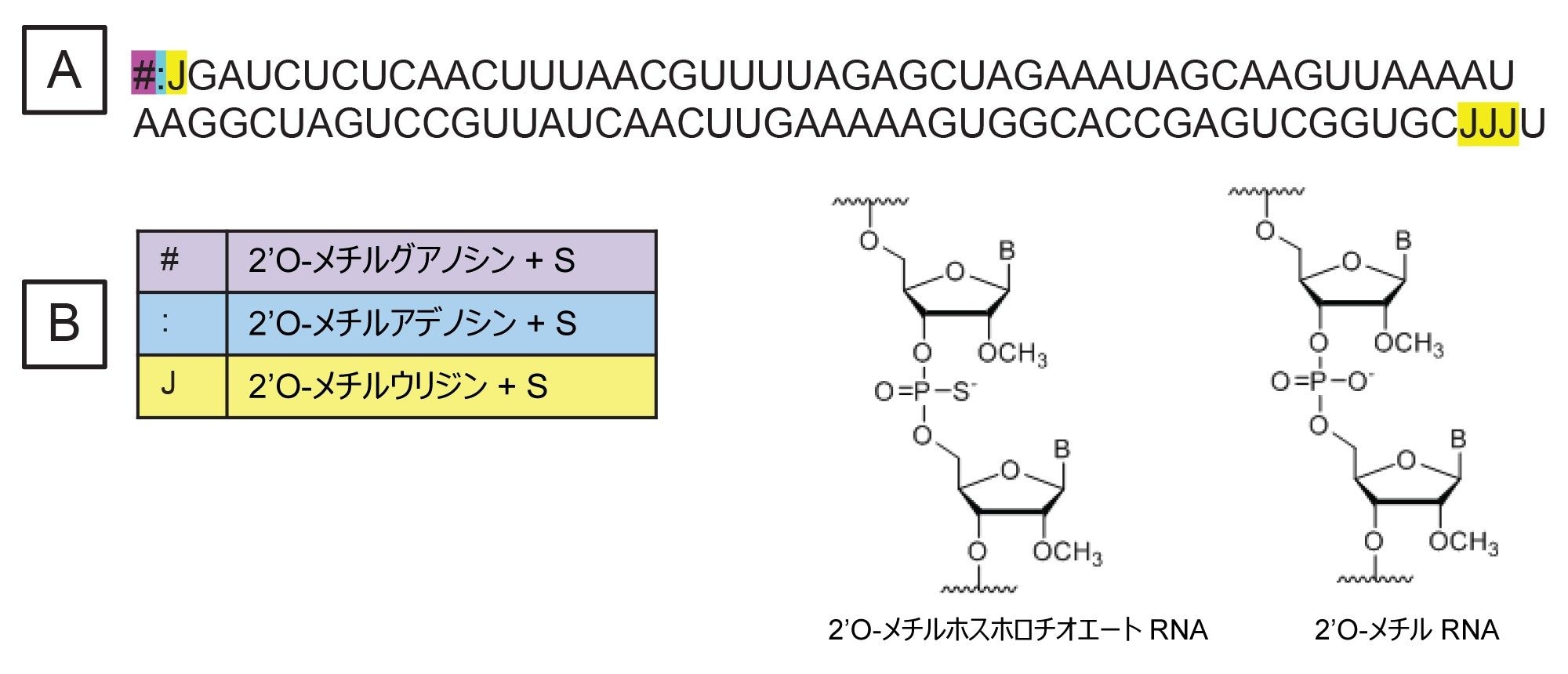 図 1. 分析した sgRNA の配列。「#」は 2'O-メチルグアノシン、「:」は 2'O-メチルアデノシン、「J」は 2'O-メチルウリジンをそれぞれ表します。すべての修飾ヌクレオチドにチオリン酸基が含まれています。
図 1. 分析した sgRNA の配列。「#」は 2'O-メチルグアノシン、「:」は 2'O-メチルアデノシン、「J」は 2'O-メチルウリジンをそれぞれ表します。すべての修飾ヌクレオチドにチオリン酸基が含まれています。
データセットはすべて、UNIFI™ アプリケーション(バージョン 3.6.0)を使用して waters_connect インフォマティクスプラットホーム(バージョン 3.2.0)内で取り込み、INTACT Mass アプリケーション(バージョン 1.6.0)を使用して解析しました。バージョン 1.0.0 の mRNA Cleaver およびバージョン 1.0.0 の Coverage Viewer マイクロアプリを使用しました。
LC 条件
|
LC-MS システム: |
ACQUITY™ Premier UPLC™(バイナリー)を搭載した BioAccord LC-MS システム |
|
プレカラム: |
VanGuard FIT カートリッジホルダー(製品番号:186007949)付きの、1.7 µm BEH™ C18 粒子を充塡した 2.1 × 5 mm ACQUITY Premier FIT カートリッジ(製品番号:186009459) |
|
カラム: |
ACQUITY Premier OST カラム 1.7 µm、130 Å、2.1 × 150 mm(製品番号:186009486) |
|
カラム温度: |
60 ℃ |
|
流速: |
300 µL/分 |
|
移動相: 移動相 A: |
8 mM DIPEA(N,N-ジイソプロピルエチルアミン)、40 mM HFIP(1,1,1,3,3,3-ヘキサフルオロイソプロパノール)含有脱イオン水(pH 8.8) |
|
溶媒 B: |
4 mM DIPEA、4 mM HFIP 含有 75% エタノール |
|
サンプル温度: |
6 ℃ |
|
サンプルバイアル: |
QuanRecovery MaxPeak HPS バイアル(製品番号:186009186) |
|
注入量: |
5 µL |
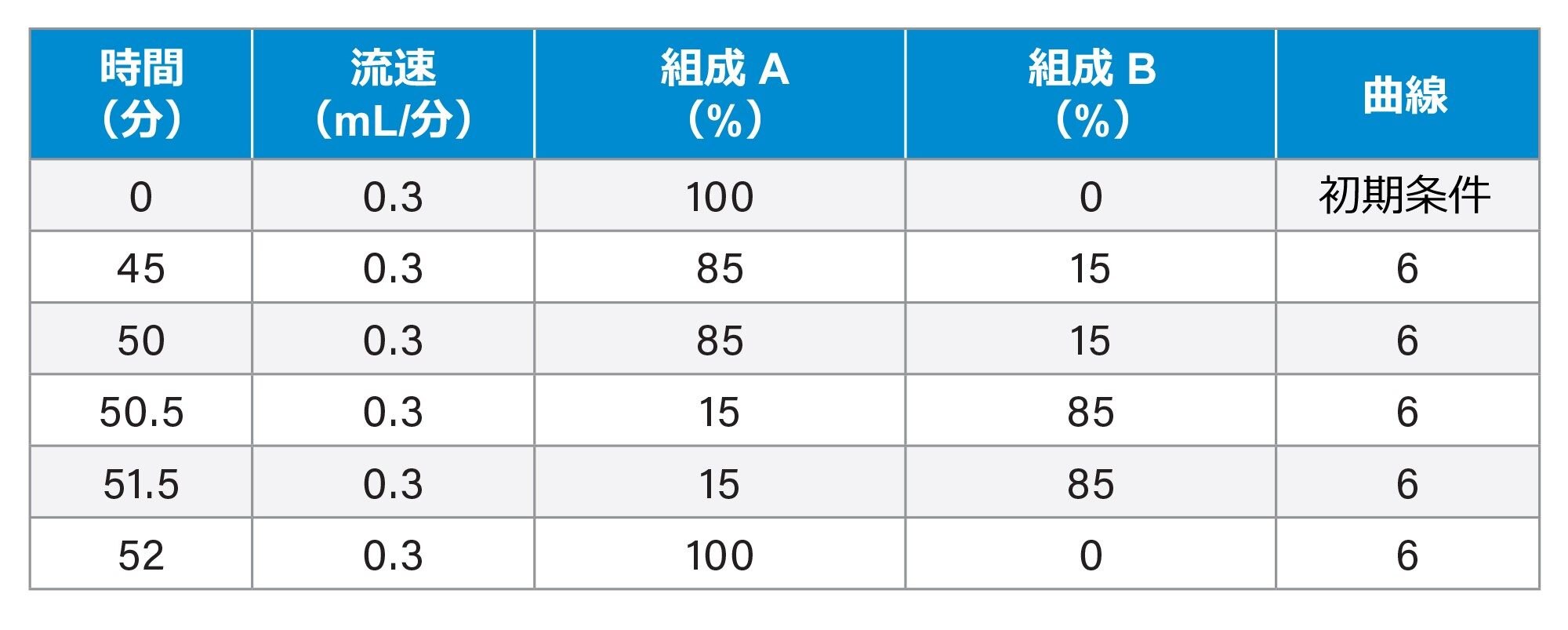
sgRNA 実験のグラジエントテーブル
MS 条件
|
取り込みモード: |
フルスキャン |
|
イオン化モード: |
ESI(-) |
|
キャピラリー電圧: |
0.8 kV |
|
コーン電圧: |
45 V |
|
イオン源温度: |
120 ℃ |
|
脱溶媒温度: |
500 ℃ |
|
脱溶媒ガス(N2)圧力: |
6.5 bar |
|
ToF 質量範囲: |
m/z 400 ~ 5000 |
|
取り込み速度: |
2 Hz |
|
ロックマス: |
waters_connect ロックマス溶液(製品番号:186009298) |
|
データ取り込みおよび解析用インフォマティクスプラットホーム |
waters_connect v.3.2.0 |
|
データ取り込み: |
UNIFI アプリ、バージョン 3.6.0 |
|
データ解析: |
INTACT Mass アプリ、バージョン 1.6.0 |
結果および考察
オリゴマッピングのデータ解析ワークフロー
核酸の CQA 分析を行う必要性の高まりに対処するために、LC-MS ベースのオリゴマッピング手法が過去数十年にわたって開発されてきました19-23。 オリゴマッピングは、酵素消化と精密質量決定によりターゲットの核酸の一次構造(配列)が得られることから、ペプチドマッピングに類似しています。オリゴマッピングで使用される一般的な酵素には、RNase T1、RNase A、MazF などがあります。今回説明する酵素消化では、すべてのグアノシンの 3' 末端の後でオリゴヌクレオチドを切断する RNase T1 を使用しました。RNase T1 のメカニズムはよく研究されており、以前に報告されています24-27。 使用した酵素が 1 種類のみであったため、100% のシーケンスカバレッジは期待しませんでした。通常、オリゴマッピング実験では複数の酵素を並行して使用します。これは、各酵素が異なる特異性を持ち、異なるセットの消化フラグメントが生じるためです。これにより、各酵素によって得られる消化フラグメントの配列がオーバーラップして、シーケンスカバレッジが高まるはずです。しかし、このアプリケーションノートの説明を簡潔にするため、ここでは 1 種類の酵素に限定しています。
オリゴマッピング実験の別の戦略として、酵素消化の条件を変更することで、オリゴヌクレオチドを意図的に部分消化するやり方があります。部分消化を使用することで、核酸の消化フラグメントの平均長が長くなるため、配列と質量の両方においてより一意になります。このアプローチでは、カバー率は向上しますが、部分消化の制御が困難であり、重複が予測される追加の m/z ピークや不完全消化フラグメントの共溶出により、すでに複雑なサンプルがさらに複雑になる可能性があります。今回示したデータでは、対象の RNA を完全に消化することが目的であり、そのために最適化した酵素条件を使用しました。
1. in silico 消化成分の生成
オリゴマッピングおよびその他の核酸の CQA のデータ解析ワークフローを図 2 に示します。in silico 消化フラグメントは、mRNA Cleaver マイクロアプリを使用して生成しました。このマイクロアプリのグラフィックユーザーインターフェース(GUI)を図 3A に示します。このソフトウェアの操作性を向上させるためのユーザーガイドが GUI の左手側にあります。mRNA Cleaver マイクロアプリは、オリゴマッピング調査のための柔軟な消化カリキュレーター兼ライブラリー作成ツールです。
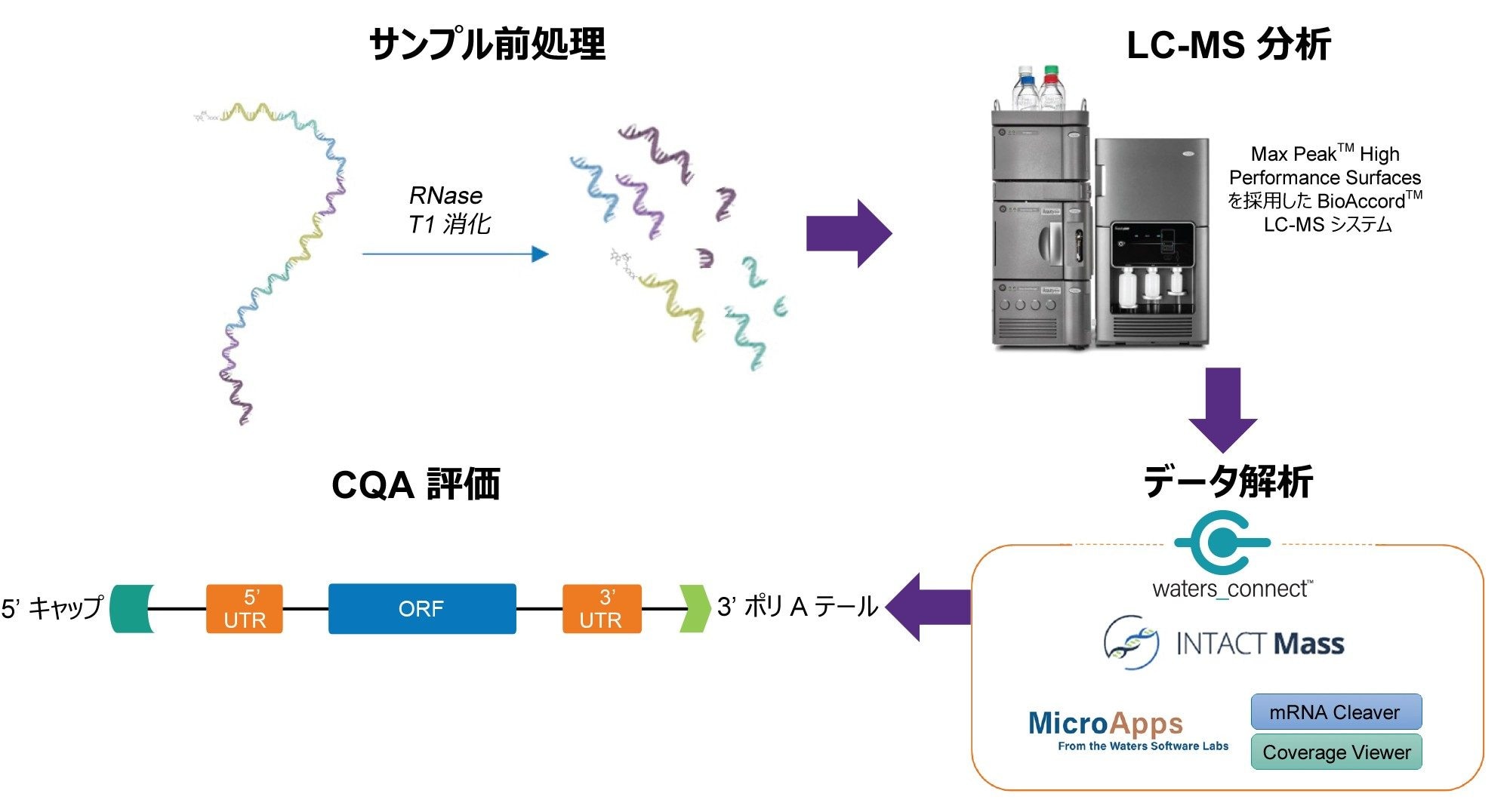 図 2. RNA の CQA の特性解析用ワークフローのスキーム
図 2. RNA の CQA の特性解析用ワークフローのスキーム
ユーザーは、3 つの埋め込みファイルを変更して、固有のニーズに合わせてカリキュレーターツールをカスタマイズすることができます。例えば、対象の配列に修飾ヌクレオチドが含まれている場合は、質量および化学組成、並びにこれらを表す 1 文字(または記号)を residues.csv ファイル(ファイルパスはユーザーガイドにあります)に追加することができます。修飾ヌクレオチドを表す文字(または記号)を使用することで、調整済み質量および化学組成を使用して関連する消化産物を算出するように、プログラムが指示されます。同様に enzymes.csv ファイルおよび modifications.csv ファイルを変更することで、ユーザーの目的に合わせて、さまざまな酵素や mRNA 固有の修飾(別の 5' キャップや 3' ポリ(A)の分布など)を追加することができます。
消化固有の設定は配列フィールドの下にあります(図 3A)。これには、消化産物の末端の調整、特定の消化産物を生成すべきかどうか、含める切れ残りの数、mRNA 固有の修飾を考慮すべきかどうかなどが含まれます。生成した消化産物のリストのフィルターが右手側にあり、ここで生成された消化産物が長さの設定に基づいてフィルタリングされます。さらに、Filter Duplicate Sequences(重複配列のフィルタリング)ボックスにチェックを入れるかどうかを選択することができます。重複配列とは、同じ配列のアイデンティティーを持つ消化フラグメントのことです。mRNA Cleaver では既定でこのボックスが選択されています。つまり、重複配列がある場合、その配列は一度だけリストされ、その配列がある別の場所は別の列にリストされます。このワークフローでは、後続のデータ分析ステップでの Coverage Viewer の使用においては、このボックスは選択していません。MS 固有の設定がフィルターの下にあり、ここでユーザーは、予想質量イオンの極性、生成されたリストでモノアイソトピック質量または平均質量を参照するかどうか、各消化産物について生成する荷電状態の範囲、生成する m/z の範囲を指定することができます。Calculate(計算)をクリックすると、消化産物のリストが生成され、このリストがカンマ区切りファイル(.csv)として出力されます。出力の例を図 3B に示します。この .csv ファイルは、ターゲットを絞った検索成分解析として INTACT Mass にインポートされます。
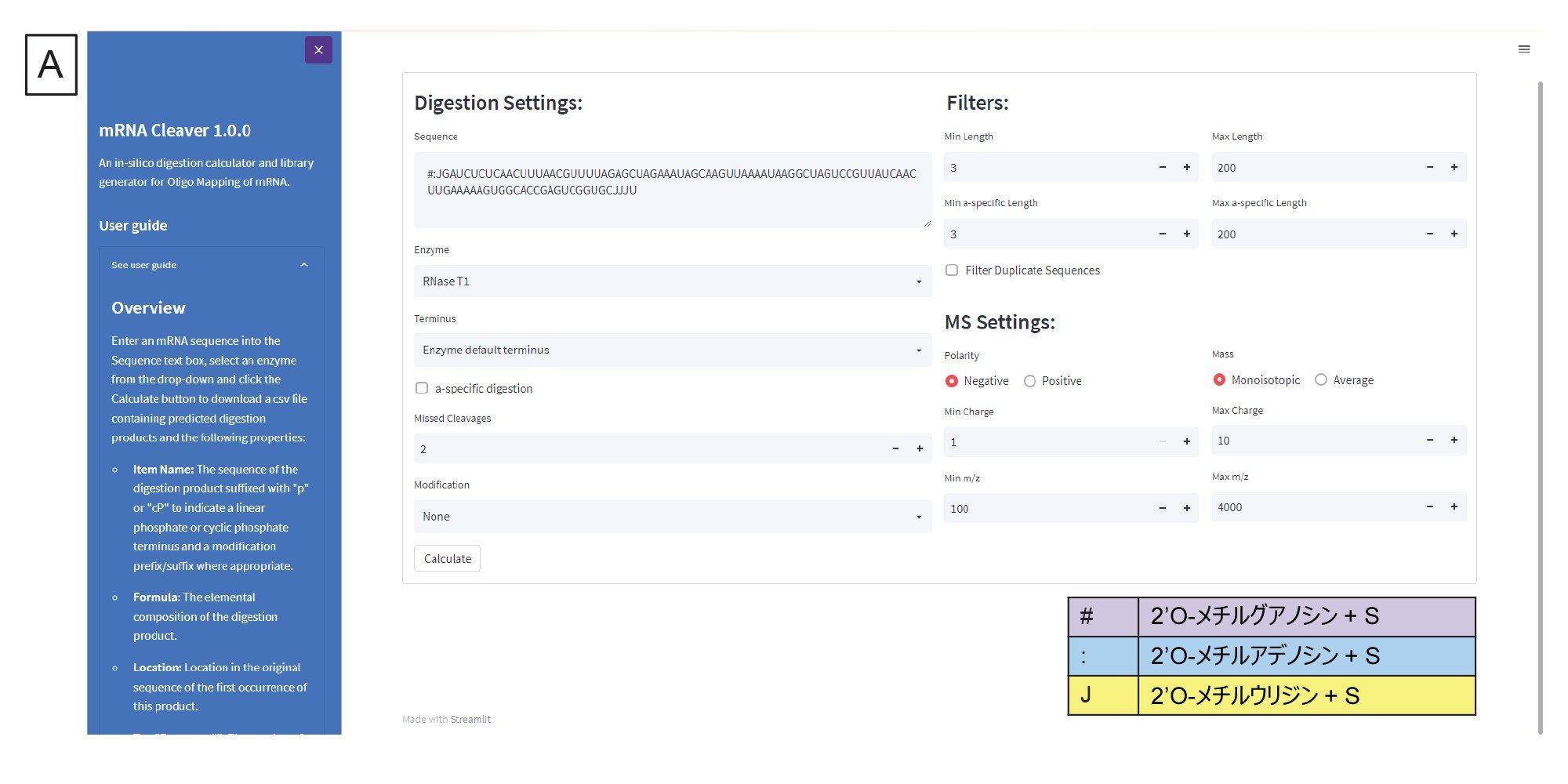 図 3.(A)mRNA Cleaver マイクロアプリのユーザーインターフェース(GUI)
図 3.(A)mRNA Cleaver マイクロアプリのユーザーインターフェース(GUI)
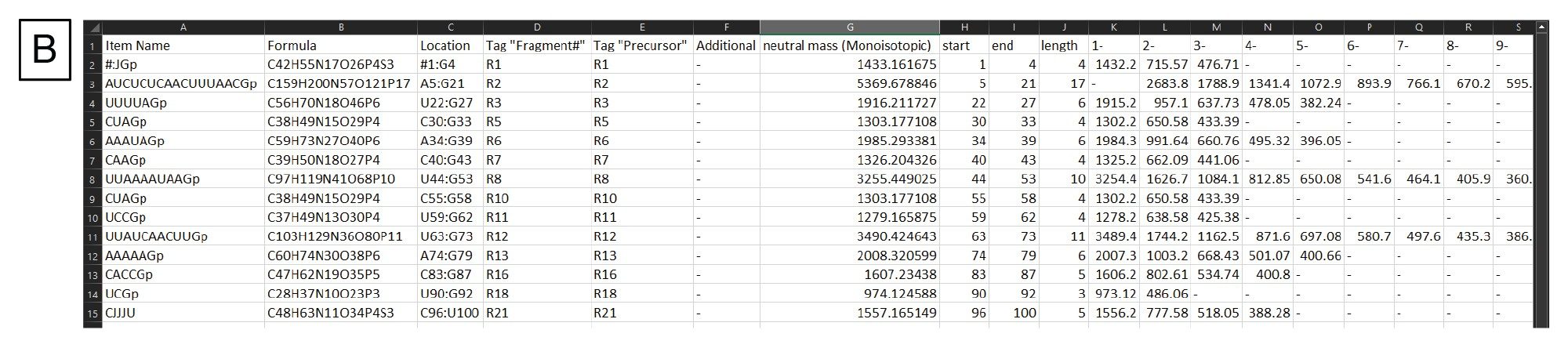 図 3.(B)mRNA Cleaver からの出力例
図 3.(B)mRNA Cleaver からの出力例
2. オリゴフラグメントの質量確認
in silico 消化産物のリストが生成され、消化済みサンプルについて実験データが収集されたら、ワークフローの次のステップは、waters_connect 内の INTACT Mass アプリを使用してデータを解析することです。mRNA Cleaver マイクロアプリからの出力により、消化産物の予想質量およびその分子 ID が得られます。これらをアプリケーション内の Expected Masses(予想質量)列に直接コピーすることができます。修飾を追加して、一般的な付加イオンおよび配列の修飾を含めることができます。図 4A には分析で解析が終了した後の結果の画面が含まれ、図 4B には代表的なデコンボリューション済みスペクトル出力が示されています。INTACT Mass で得られる結果、MS、UV、TIC の各テーブルは、テーブルの右手側にあるダウンロードボタンを使用してダウンロードすることができます。
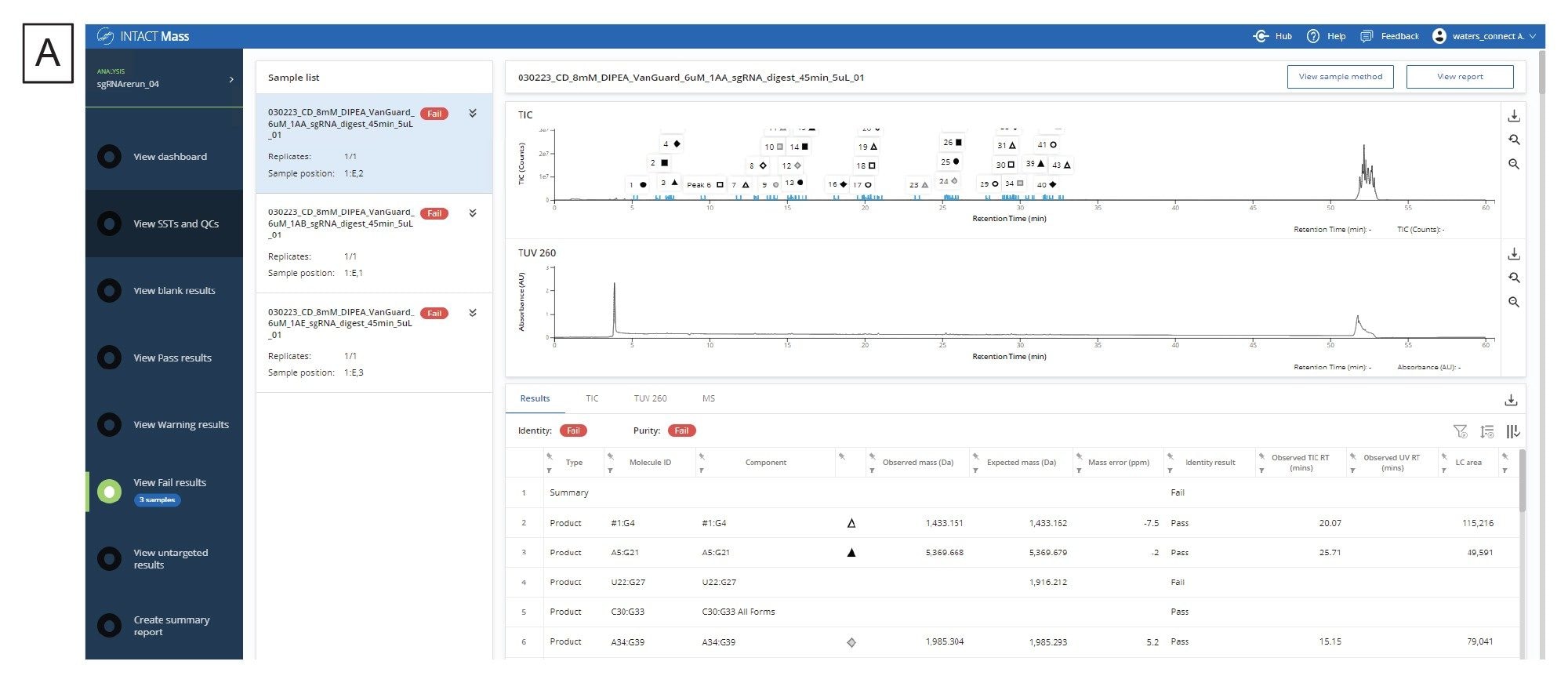 図 4.(A)INTACT Mass アプリケーションの結果のダッシュボード
図 4.(A)INTACT Mass アプリケーションの結果のダッシュボード
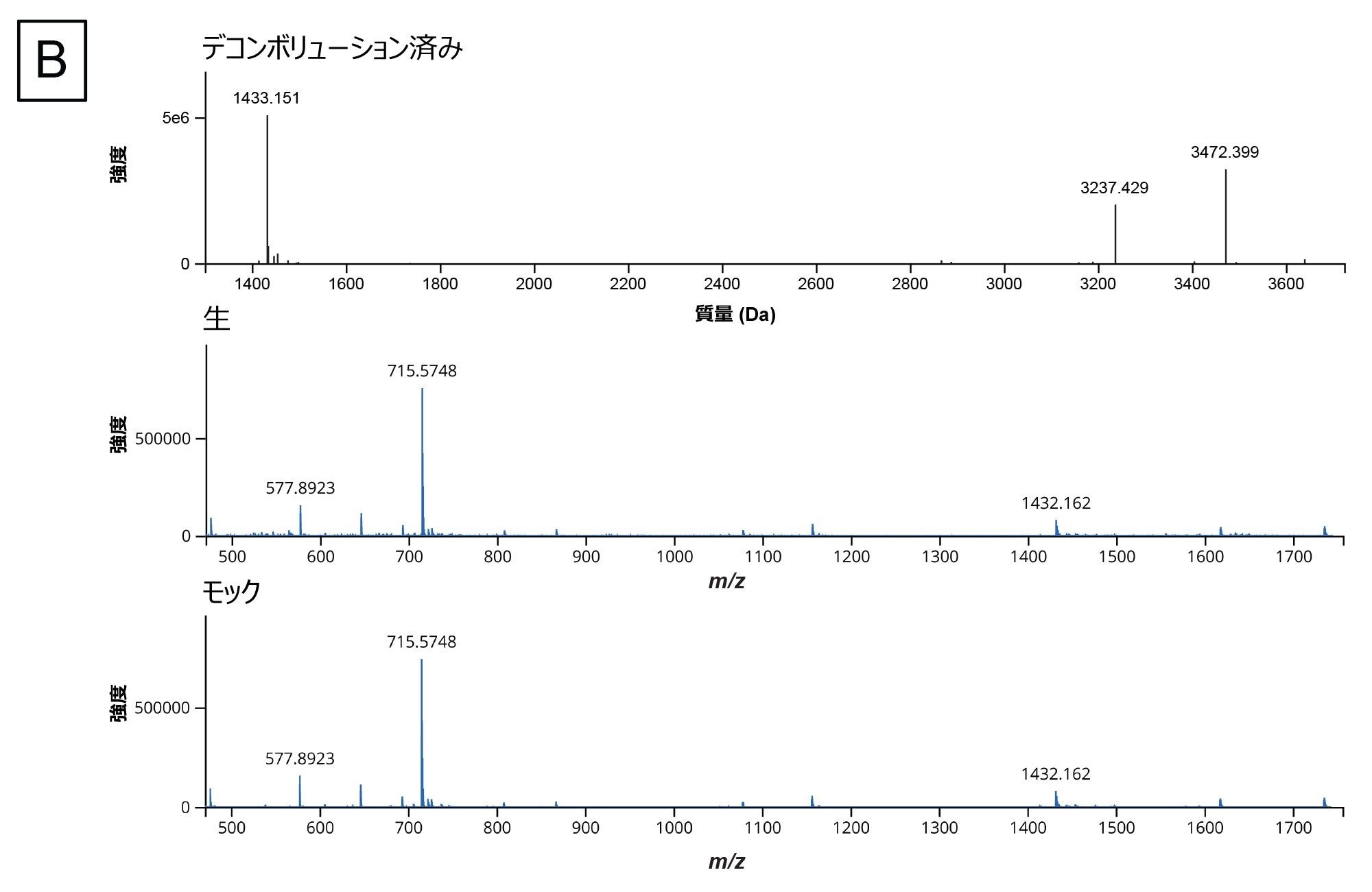 図 4.(B)デコンボリューションしたスペクトルの出力例
図 4.(B)デコンボリューションしたスペクトルの出力例
INTACT Mass アプリによって生成された表形式のデータは、フラグメントのマッピングデータを示す重要な結果ですが、Coverage Viewer マイクロアプリを使用して、配列に対するこれらの結果の視覚化およびシーケンスカバレッジの計算を行うことができます。Coverage Viewer マイクロアプリの GUI を図 5 に示します。対象の RNA 分子の配列が配列フィールドに入力されており、ワークフローの最初のステップで生成された mRNA Cleaver の出力をインポートして、ターゲット消化フラグメントリストに入力することができます(左下隅)。左側の Import(インポート)ボタンを使用して INTACT Mass の結果ファイルをインポートすることができます。ターゲットが確認されると、シーケンスカバレッジが計算され、GUI の右手側に表示されます。INTACT Mass の結果ファイルをインポートすると、キーワード Pass(合格)が付いた消化フラグメントは Confirmed(確認)と見なされます。ユーザーがチェックを外すことで、手動で割り当てた産物も Confirmed(確認)と見なされるようにすることができます。関連する配列の下の赤色の領域が緑色に変わり、配列の特定の部分が精密質量測定によって確認されたことが示されます。完了後は、Confirmed(確認)列のターゲットとする消化フラグメントの部分を .csv としてエクスポートし、後で使用することができます。さらに、Capture image to clipboard(画像をクリップボードにキャプチャー)をクリックしてカバレッジマップの画像をコピーし、別のプログラムに保存することができます。
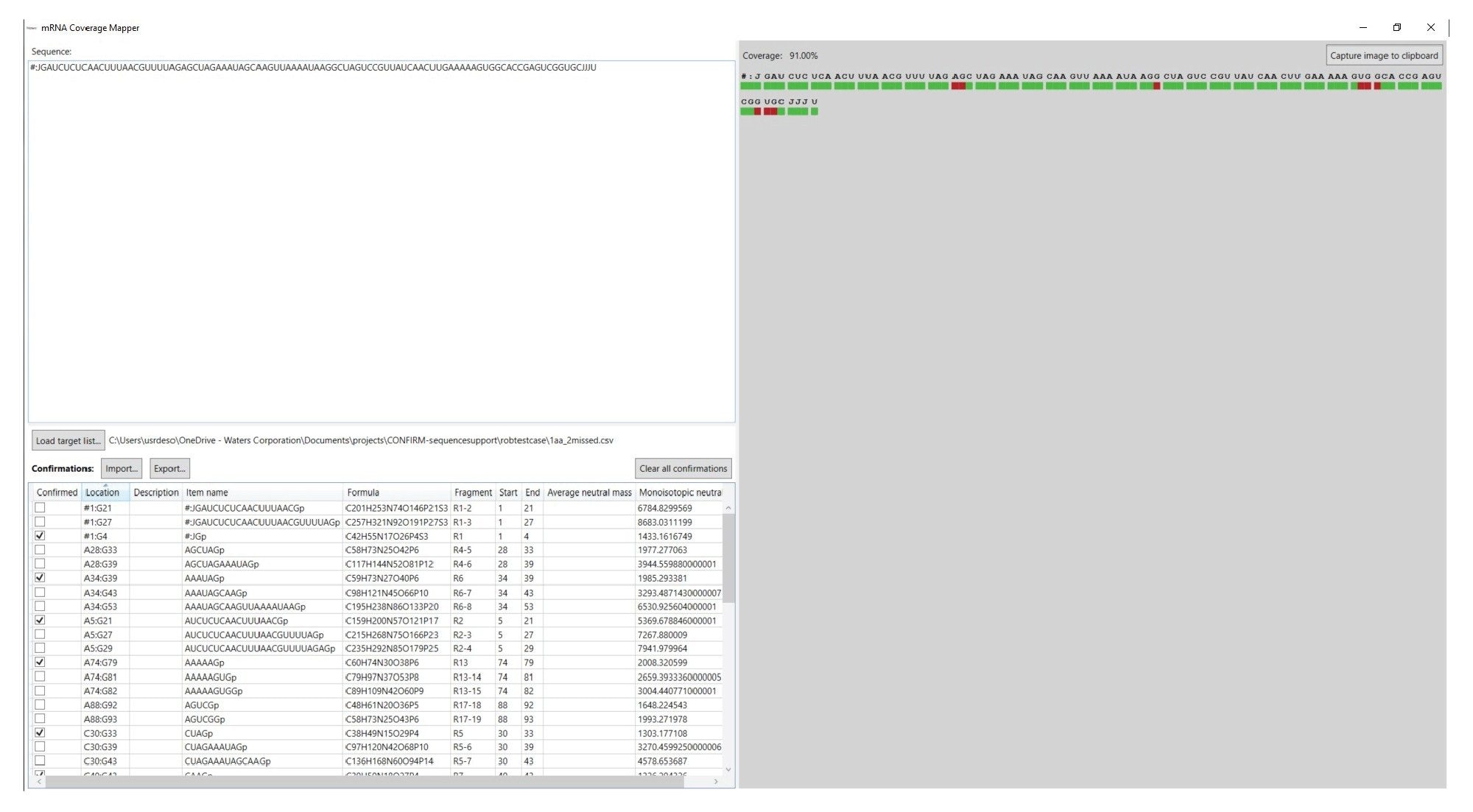 図 5. Coverage Viewer マイクロアプリのユーザーインターフェース
図 5. Coverage Viewer マイクロアプリのユーザーインターフェース
3. テストケース:sgRNA の消化およびマッピング
CRISPR ベースの遺伝子編集プロセスはシンプルであることから、ここ数年で一般的になっています16。このプロセスでは、ガイド RNA と CRISPR 関連(Cas)ヌクレアーゼという 2 つの主要成分を使用します16。ガイド RNA は、対象のターゲット DNA を認識し、Cas ヌクレアーゼを正しい編集部位に導く特異的 RNA 配列です16。Cas ヌクレアーゼは、特定の DNA 部位に作用するガイド RNA に完全に依存する非特異的ヌクレアーゼです16。元々、ガイド RNA は、ターゲットに相補的な Crispr RNA と Cas ヌクレアーゼの結合スキャフォールドとして機能する tracr RNA という 2 つの別々のオリゴヌクレオチドで構成されていました16。テクノロジーが進化して、crisper RNA と tracr RNA は「リンカーループ」と呼ばれる領域で 1 つに融合され、シングルガイド RNA(または sgRNA)になりました16。合成で生成された sgRNA の重要な CQA は、分子の特定の部位に組み込まれた修飾残基を含む、その配列の確認です。
ワークフローを使用して RNase T1 消化を行い、消化産物を分析することで、消化産物を同定し、MS1 レベルで(追加の MS フラグメンテーションを行うことなく)、sgRNA 配列のマッピングカバレッジを確認することができます。まず、sgRNA の修飾ヌクレオチドを residuals.csv ファイルに追加して、mRNA Cleaver が sgRNA のin silico 消化産物を正しく決定できるようにしました。次に、mRNA Cleaver マイクロアプリの消化および MS の設定を調整して実験条件を反映するようにしました。具体的には、使用する酵素が RNase T1 であり、mRNA Cleaver が 2 箇所の切れ残りを含む消化フラグメントを生成するように設定しました。完全消化であっても、切れ残りの変数を 2 に増やすことで、酵素がすべての G で切断しない可能性があるため、この可能性を考慮します。次に、in silico 消化産物のリストを生成し、.csv ファイルにエクスポートしました。2 つの非特異的な配列(配列 CUAG を持つ 4 mer のオリゴ)が同定されました。非特異的な配列は、消化フラグメントで、異性体および/または同重体です。このことは、これらの配列の m/z ピークおよび荷電状態エンベロープがオーバーラップしていることを意味します。分析において重要な RNA の一部が含まれる非特異的な配列には、気相でのフラグメンテーションなど、配列のアイデンティティーを確認するためのさらなる実験的ステップが必要になる場合があります。一方、その他の 19 種の消化フラグメントには、マススペクトルで区別できるはずの固有の予想質量および配列のアイデンティティーがあります。この仮定は、BioAccord によって得られた予想質量の精度、および mRNA Cleaver マイクロアプリで得られた各消化フラグメントの> in silico で生成された m/z の範囲に基づいています。
実験方法の記載に従って in vitro 消化を行い、消化済み sgRNA および消化されていないインタクト sgRNA のデータを両方、BioAccord LC-MS システムで waters_connect を使用して収集しました。図 6A に、インタクト sgRNA および後続の消化済み sgRNA のトータルイオンクロマトグラムを示します。消化済みフラグメントはグラジエントの 5 ~ 40 分の間に溶出しています。sgRNA は完全に消化されています。それは、インタクト sgRNA は 35.6 分で溶出しており、消化サンプルのトレースにはこれに対応するピークがないことからわかります。
LC-MS データと in silico 消化成分のリストの両方が INTACT Mass アプリに送信され、一致の許容誤差 10 ppm の質量マッチに基づいてさまざまな消化成分が割り当てられました。次に、INTACT Mass の結果を Coverage Viewer のマイクロアプリにインポートしてシーケンスカバレッジを視覚化しました(図 6B)。その結果、MS1 シーケンスカバレッジ 91% が得られました。このテストケースでは、非特異的な配列の配列アイデンティティーが同じであり、配列アイデンティティーを確認するための衝突誘起フラグメンテーションは不要になるため、行いませんでした。ただし、非特異的な配列が多い場合や対象の非特異的な配列の配列アイデンティティーが異なる場合は、これらの非特異的な配列の配列アイデンティティーがフラグメンテーションによって確認できない限り、非特異的な配列をシーケンスカバレッジの計算から除外することもできます28。シーケンスカバレッジ 100% を達成するため、RNase T1 とは異なる切断特異性を示す MasF や RNase A などの別のエンドリボヌクレアーゼを使用して、sgRNA 配列の未同定部分をカバーすることもできます。
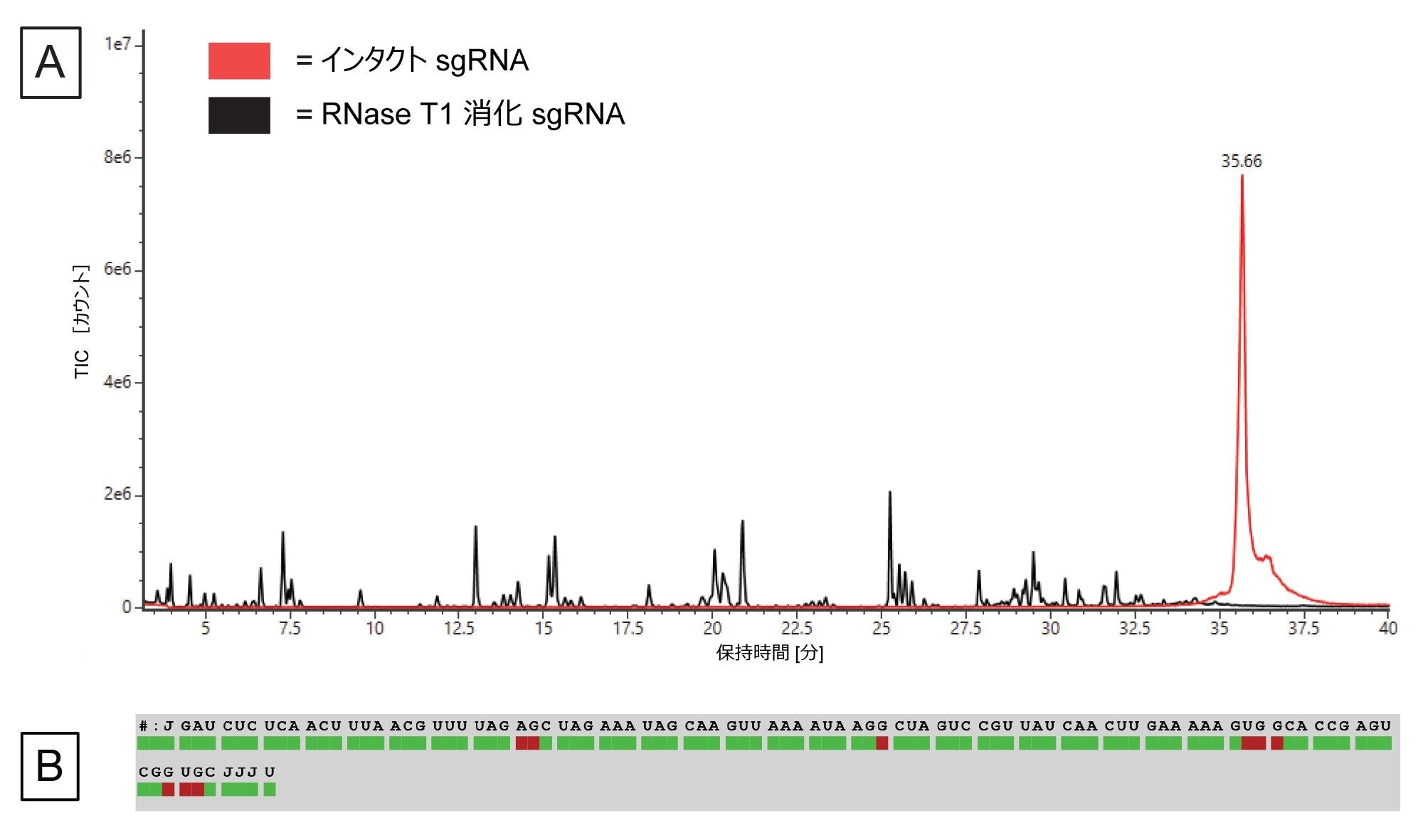 図 6.(A)消化済み sgRNA の TIC(黒色のトレース)および消化されていないインタクト sgRNA(赤色のトレース)の TIC。(B)Coverage Viewer マイクロアプリで視覚化した、LC-MS によって得られたシーケンスカバレッジ。
図 6.(A)消化済み sgRNA の TIC(黒色のトレース)および消化されていないインタクト sgRNA(赤色のトレース)の TIC。(B)Coverage Viewer マイクロアプリで視覚化した、LC-MS によって得られたシーケンスカバレッジ。
mRNA の消化およびマッピング
このワークフローは、メッセンジャー RNA(mRNA)などのより大きな RNA にも適用できます。ただし、大きな RNA を MS1 レベルでマッピングすることは、追加で多くの非特異的な配列と共溶出する分子種が存在し、データの複雑さが指数関数的に増加するため、大掛かりな作業になります。Gaye らは、IP-RP LC-MS および waters_connect プラットホームを使用して mRNA 配列のマッピングを最適に行うための方法に関するアプリケーションノートを以前に公表しています18。今回、更新したワークフローを使用してデータを再解析しました。
図 7A に、Gaye らから転載した RNase T1 mRNA 消化物の TIC クロマトグラムおよび Coverage Viewer の結果を示します18。挿入図は、8.58 分に溶出するピークからの抽出質量スペクトル(XIC)です。この代表的な XIC では、複数の分子種がピークの下で共溶出しており、データの複雑さが浮き彫りになっています。このワークフローに従い、mRNA Cleaver マイクロアプリを使用して、2 つの in silico 酵素消化産物のリスト(1 つは非特異的な配列を含み、1 つは含まない)を作成しました。これらのリストおよび取り込まれた LC-MS データを INTACT Mass アプリに送信し、データ解析を行いました。次に、INTACT Mass の結果を Coverage Viewer マイクロアプリにインポートしました。
次に、INTACT Mass の結果を Coverage Viewer マイクロアプリにインポートしました。MS1 レベルの消化フラグメントマップから、一意のシーケンスカバレッジは 38.87%、全体的なシーケンスカバレッジ(非特異的な配列は含まれていません)。これらのマップをそれぞれ図 7B および図 7C に示しています。 このシーケンスカバレッジは、別のエンドリボヌクレアーゼを並行して使用することで改善できますが、データの複雑さ(共溶出する分子種、非特異的な配列など)が、データを解釈する上で引き続き課題となります。このような複雑なサンプルでは、データの解析にかかる時間が分析するピークの数とともに長くなり、分析のスループットを制限するようになる可能性があります。
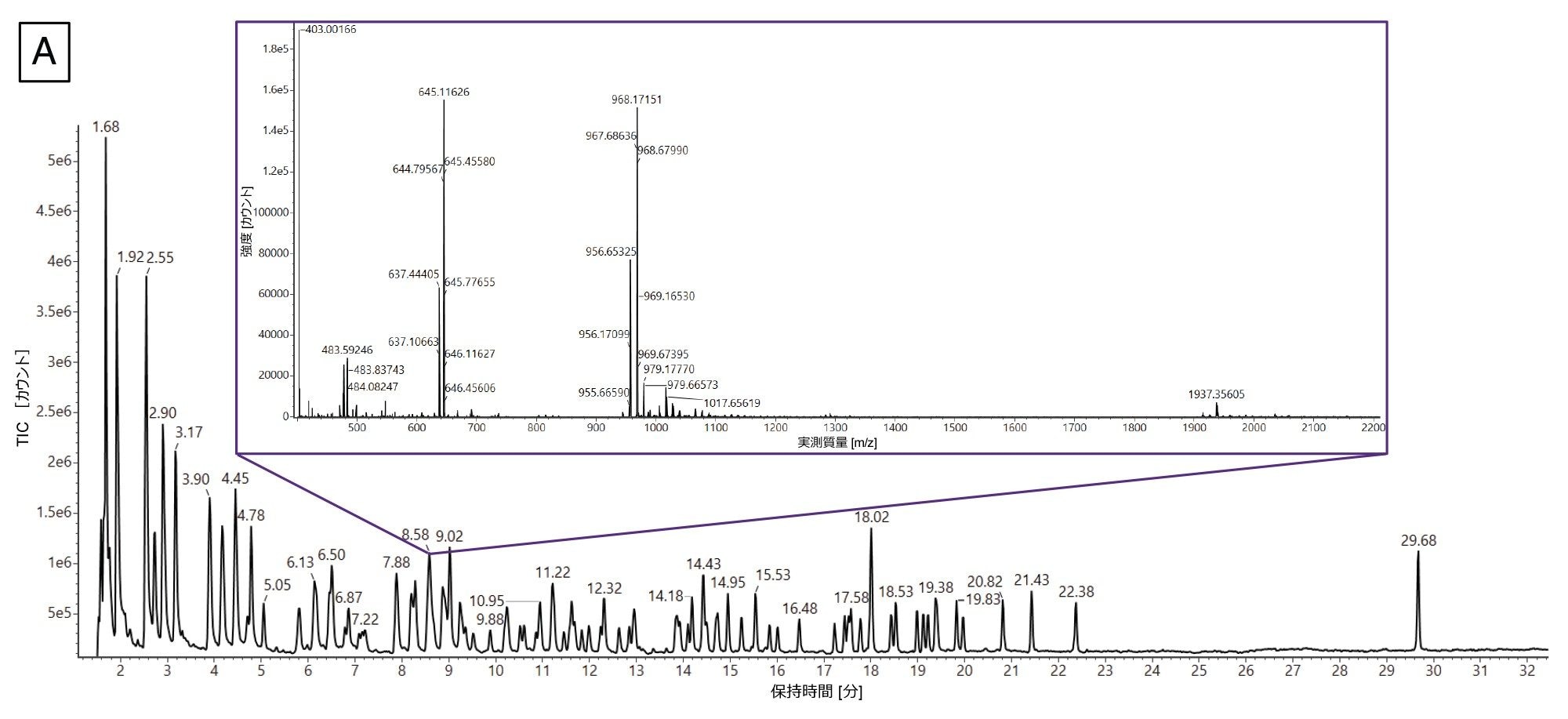 図 7.(A)Gaye らから転載した mRNA 消化物の TIC(18)。挿入されているスペクトルは、共溶出する複数のオリゴヌクレオチド分子種が含まれている 8.58 分に溶出するピークから得られた代表的な XIC です。
図 7.(A)Gaye らから転載した mRNA 消化物の TIC(18)。挿入されているスペクトルは、共溶出する複数のオリゴヌクレオチド分子種が含まれている 8.58 分に溶出するピークから得られた代表的な XIC です。
 図 7.(B)Coverage Viewer マイクロアプリで可視化した、MS1 によって可能になった一意のシーケンスカバレッジ(非特異的な配列が含まれていない)
図 7.(B)Coverage Viewer マイクロアプリで可視化した、MS1 によって可能になった一意のシーケンスカバレッジ(非特異的な配列が含まれていない)
 図 7.(C)Coverage Viewer マイクロアプリで可視化した、MS1 によって可能になった全体的なシーケンスカバレッジ
図 7.(C)Coverage Viewer マイクロアプリで可視化した、MS1 によって可能になった全体的なシーケンスカバレッジ
BioAccord でのマッピング実験の一環としてデータ非依存的(MSE)フラグメンテーションデータを収集する場合は、CONFIRM Sequence アプリを使用して、非特異的なフラグメントのさらなる配列解析を行うことができます。このタイプの分析の例を Gaye らから転載して図 8 に示します18。 この追加の段階の分析に関するワークフローの自動化が進行中です。
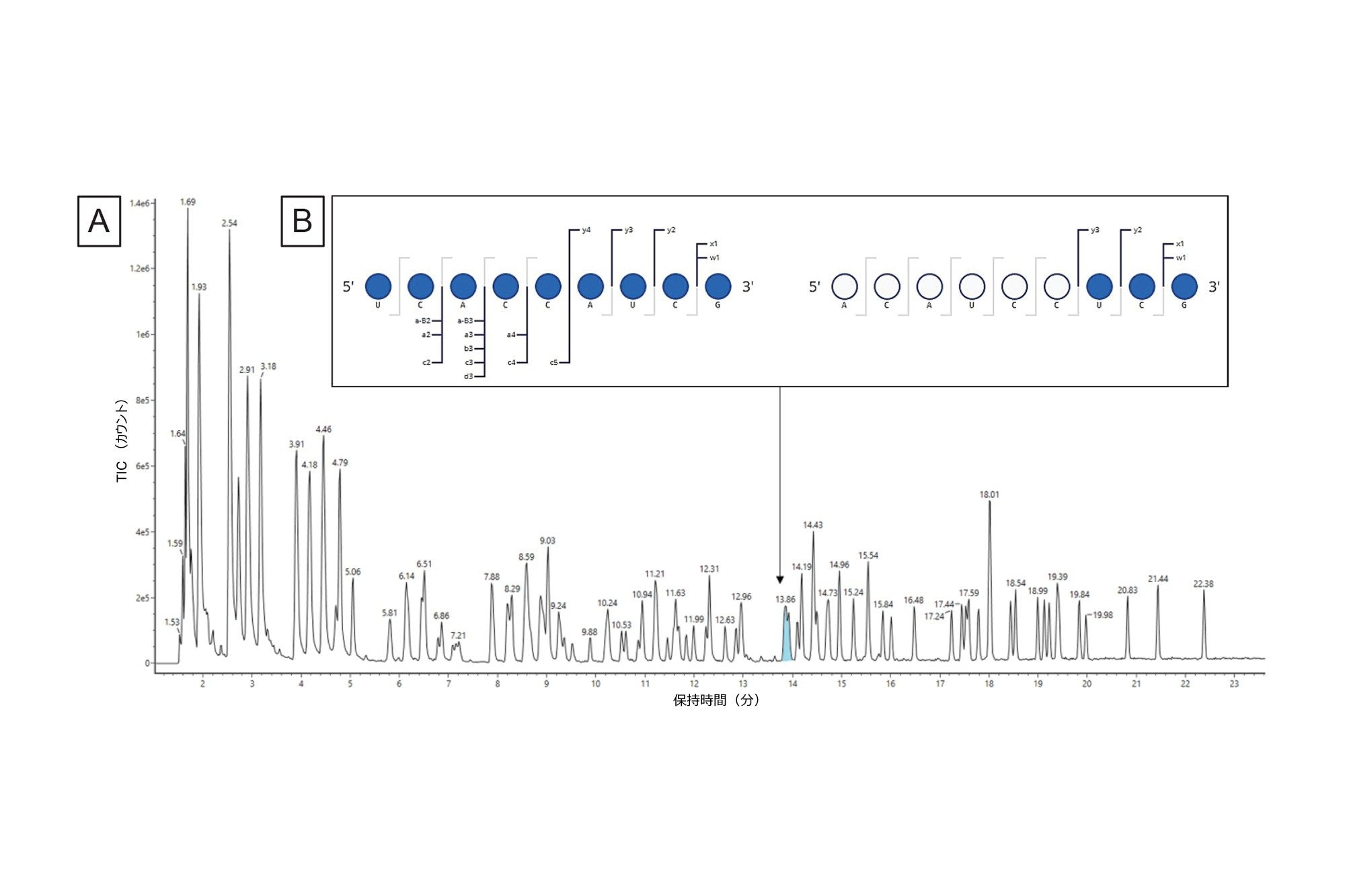 図 8.(A)位置 623 ~ 631(ACAUCCUCGp)および 551 ~ 559(UCACCAUCGp)に消化フラグメント成分が予測され、Gaye らから転載した TIC 中の同じ RT のピークに割り当てられています。インタクト質量情報を使用して正しい割り当てを決定することはできません。(B)同じ注入で得られた MSE データを使用することで、この割り当ての正しい配列を明らかにすることができます。waters_connect CONFIRM Sequence アプリケーションを用い、各配列に対して高エネルギーフラグメントイオンを予測し、カスタマイズされたアルゴリズムで波形解析した生データの同位体クラスターにマッチングしました。このソフトウェアにより、確認済みのフラグメントイオンがドットマップに表示され、シーケンスカバレッジが迅速に評価されます。
図 8.(A)位置 623 ~ 631(ACAUCCUCGp)および 551 ~ 559(UCACCAUCGp)に消化フラグメント成分が予測され、Gaye らから転載した TIC 中の同じ RT のピークに割り当てられています。インタクト質量情報を使用して正しい割り当てを決定することはできません。(B)同じ注入で得られた MSE データを使用することで、この割り当ての正しい配列を明らかにすることができます。waters_connect CONFIRM Sequence アプリケーションを用い、各配列に対して高エネルギーフラグメントイオンを予測し、カスタマイズされたアルゴリズムで波形解析した生データの同位体クラスターにマッチングしました。このソフトウェアにより、確認済みのフラグメントイオンがドットマップに表示され、シーケンスカバレッジが迅速に評価されます。
5' キャッピングの有効性の測定
mRNA に固有の CQA は、5' キャッピングの効率および構造を評価することです。mRNA 分子の 5' キャップは、核外輸送の制御、エキソヌクレアーゼによる分解の防止、翻訳の促進、5' に近いイントロン除去の促進などのいくつかの生物学的機能に不可欠です14。以前のアプリケーションノートで Nguen らは、BioAccord LC-MS システムを使用した合成 mRNA キャップ構造の迅速分析法(5 分以内)について報告しています14。
図 9A に示すように、ワークフローを使用して、デコンボリューションしたスペクトルに基づいて 5' キャップ不純物を検出し、割り当てることができました。このデータを分析するため、INTACT Mass アプリの(バージョン 1.6.0 のリリース時点における)新機能であるカスタムチャージデコンボリューションを適用しました。自動デコンボリューションでは、ピーク検出およびデコンボリューション解析向けに最適化された設定を使用しており、複雑さがさまざまなレベルの多くのサンプルのルーチン分析を簡素化するのに適しています。ただし、このデータでは分析種のダイナミックレンジが大きく、頑健な同定を行いたい分析の検出下限近くに複数の低強度の不純物が含まれているため、この場合は、自動デコンボリューションアルゴリズムの設定は適さないことが判明しました。カスタムデコンボリューションを使用することで、スペクトルのデコンボリューション設定を最適化し、分子に存在する 5' キャップ構造の完全な集団をより適切に表すことができます。
このデータでは、BayesSpray チャージデコンボリューションアルゴリズムを適用して、保持時間の範囲、入力した m/z の範囲、予想される出力質量の範囲、チャージの範囲をデータに合わせてカスタマイズしました。5' キャップ分析の INTACT Mass データを図 9B に示します。1:10 希釈サンプル中の不純物(ppG、pppG、GpppG、m7GpppG(Cap-0))をすべて同定し、Ngyuen らからの 1:100 希釈サンプル中に前述の 3 種類の不純物(pppG 以外)を同定することができました14。
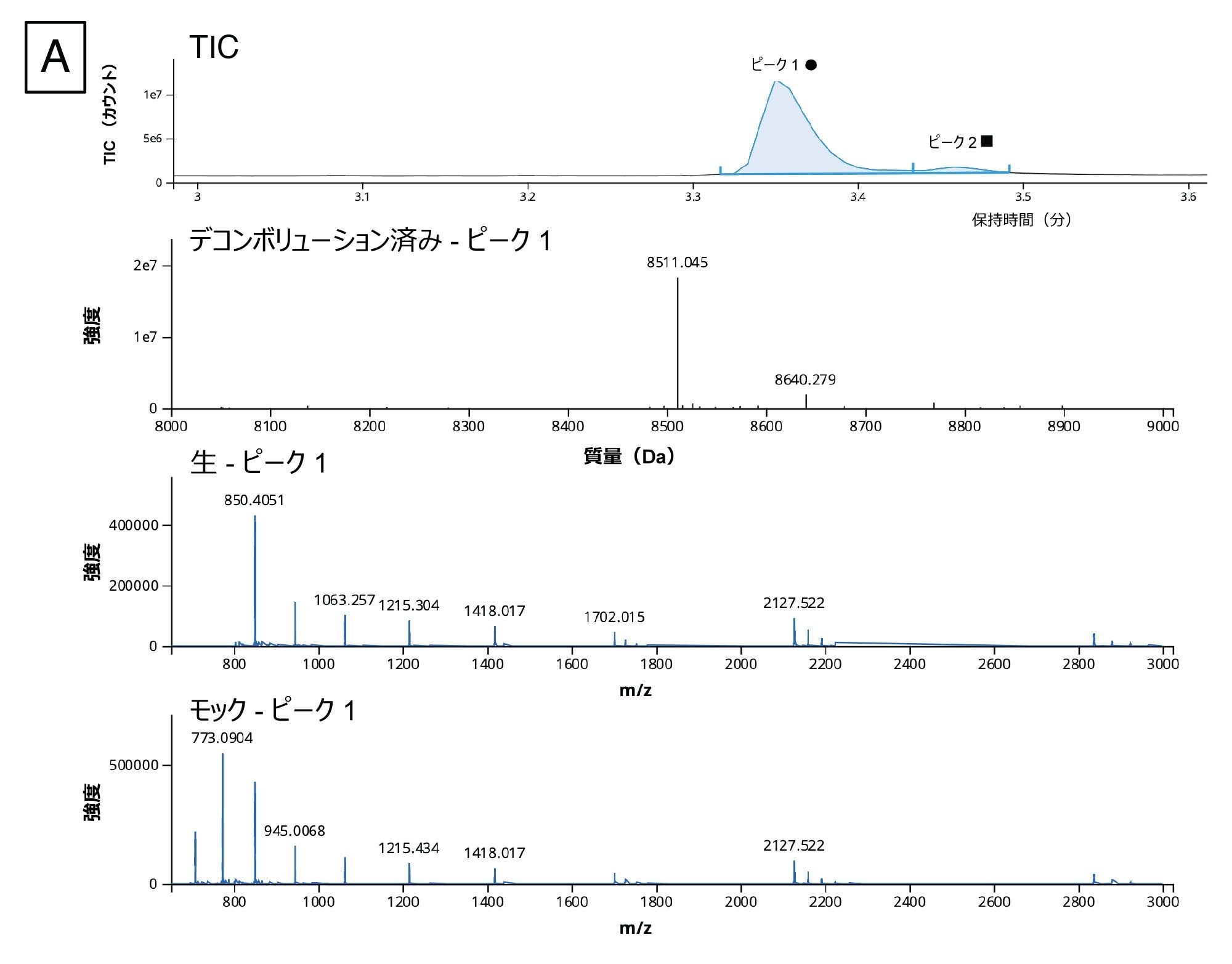 図 9.(A)3.37 分(丸印)のピークの TIC スペクトル、デコンボリューション済みスペクトル、生スペクトル、およびモックスペクトル
図 9.(A)3.37 分(丸印)のピークの TIC スペクトル、デコンボリューション済みスペクトル、生スペクトル、およびモックスペクトル
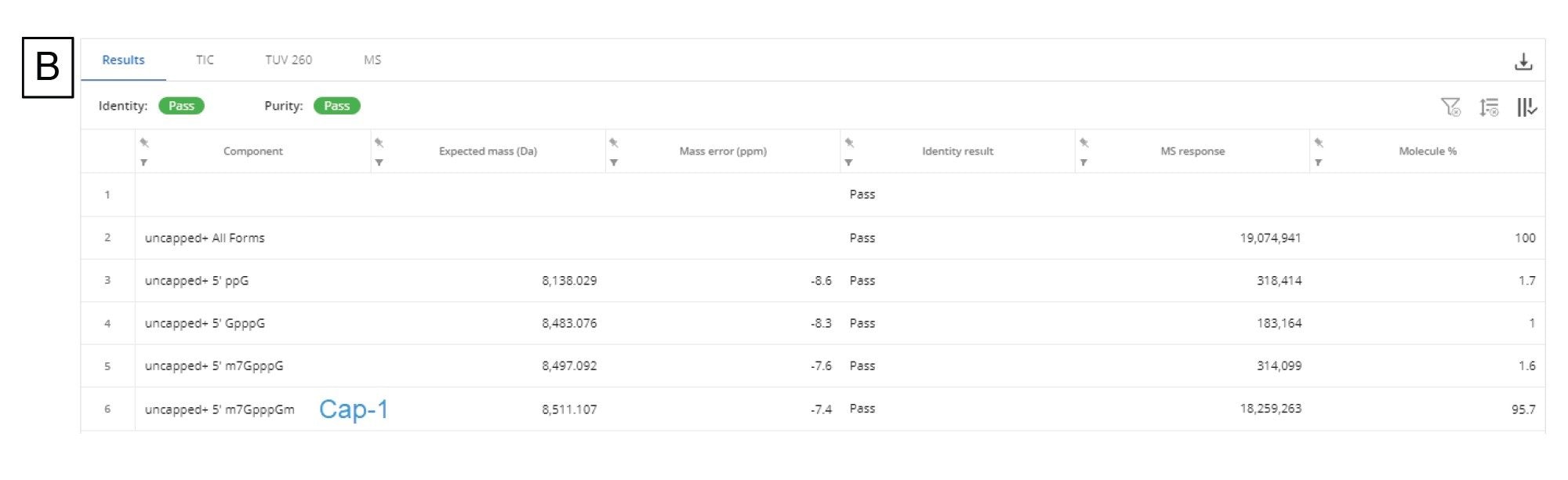 図 9.(B)Cap-1 フラグメントおよびその生成物関連の不純物フラグメントに関する INTACT Mass アプリの結果テーブル
図 9.(B)Cap-1 フラグメントおよびその生成物関連の不純物フラグメントに関する INTACT Mass アプリの結果テーブル
効果的な翻訳は mRNA の 5' キャップに大きく依存するため、正しくキャップされた mRNA 生成物の割合は、mRNA 医薬品の製造において綿密にモニターされる CQA です。Nguen らによって開発されたアッセイおよびカスタムデコンボリューション設定を使用したデータ分析ワークフローには、キャッピング過程が不完全である場合に生じる望ましくない不純物をルーチンに同定するのに十分な感度と頑健性があります14。
信頼性の高い 3’ ポリ(A)テールの長さおよび分子量分布の測定
mRNA に固有の別の CQA として、3' ポリ(A)テールの長さの決定があります。3' ポリ(A)テールの長さは、mRNA の安定性と直接相関しています17。 したがって、長さ、あるいはむしろ平均長およびサイズ分布を決定することによって、ポリアデニル化のプロセスに関する有用な情報を得ることができます。ホタルルシフェラーゼ(FLuc)mRNA の RNase T1 消化後に得られる 3' ポリ(A)テールオリゴヌクレオチドの分析用に、比較的迅速(実行時間 15 分)な LC-MS アッセイが Doneanu らによって開発されました13。図 10A に示すように、この 3' ポリ(A)テールは、TIC において、十分に分離した、遅く溶出する大きなクロマトグラフィーピークとして溶出しています。共溶出する 3' ポリ(A)分子種の集団を含む対応する ESI-MS スペクトルの組み合わせをワークフローに送信し、INTACT Mass アプリで使用可能な MaxEnt1 チャージデコンボリューションを使用してデコンボリューションしました。入力 m/z の範囲、出力質量の範囲、保持時間の範囲などのカスタムチャージデコンボリューションのパラメーターを調整して、図 10A の挿入図に示すデコンボリューション済みスペクトルを得ました。このスペクトルは、3' ポリ(A)の不均一性が大きいことを示しており、オリゴヌクレオチドのサイズは 122 ~ 132 mer で、特徴的な 329.2059 Da のアデノシン一リン酸残基分、それぞれ質量が異なります。INTACT Mass の結果を図 10B に示します。検出されたすべてのオリゴ成分の ESI-MS シグナルおよびそれらに対応する強度を、図 10C に示すプロットにまとめています。このグラフを使用して、FLuc mRNA の 3' ポリ(A)テールの重量平均質量を算出しました。平均質量の測定値は 126.5 mer の長さに対応しており、この測定値は直交する SEC の測定値から確認されました15。INTACT Mass 解析の後、11 種類の 3' ポリ(A)バリアントを、通常 45 ppm を上回る質量精度で測定しました(図 10B)。
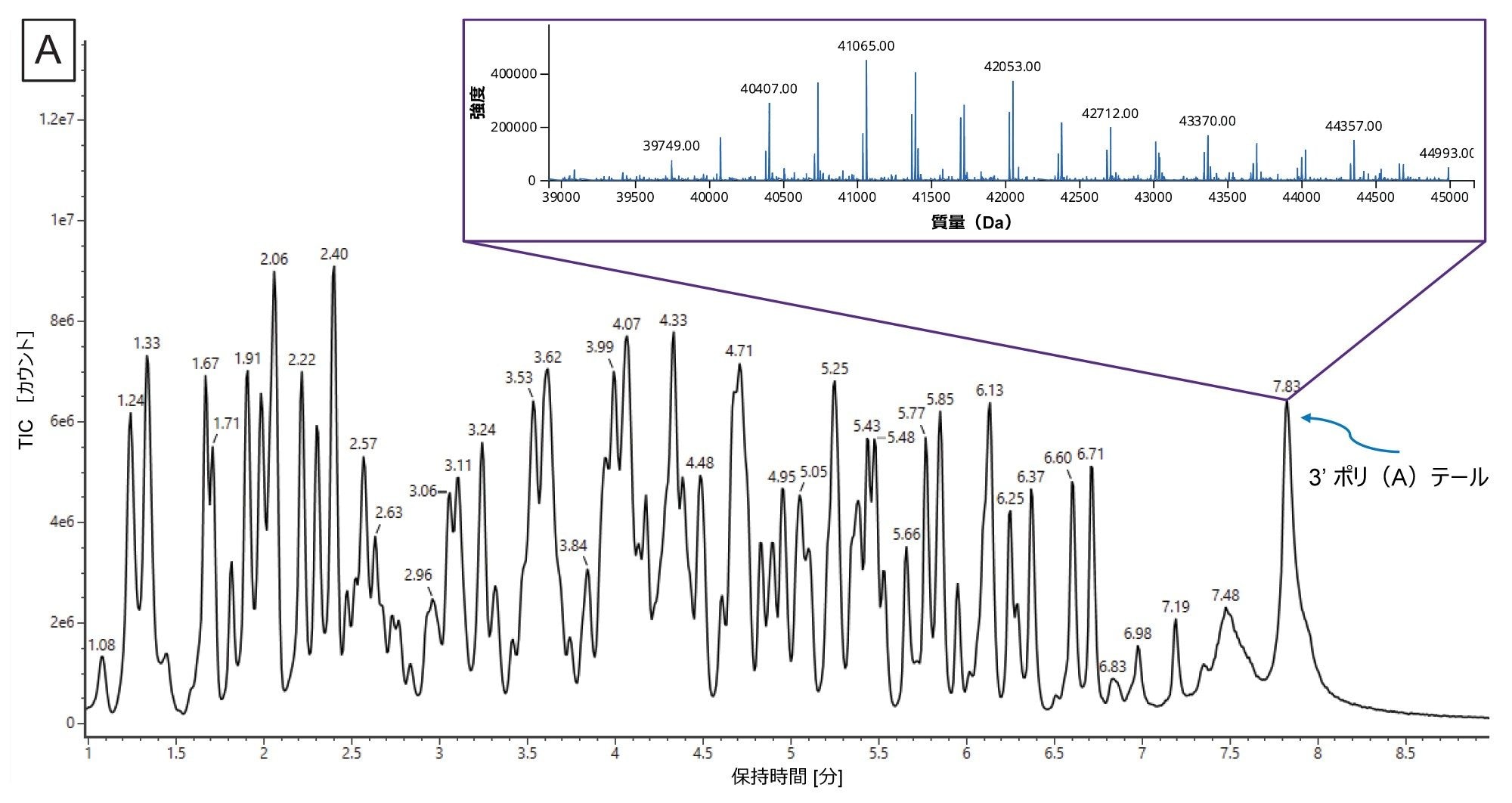 図 10.(A)RNase T1 消化 FLuc mRNA に由来する 3' ポリ(A)テールのオリゴヌクレオチド混合物の TIC クロマトグラム。挿入図に、INTACT Mass アプリケーションで得られた MaxEnt1 チャージデコンボリューションを示しています。
図 10.(A)RNase T1 消化 FLuc mRNA に由来する 3' ポリ(A)テールのオリゴヌクレオチド混合物の TIC クロマトグラム。挿入図に、INTACT Mass アプリケーションで得られた MaxEnt1 チャージデコンボリューションを示しています。
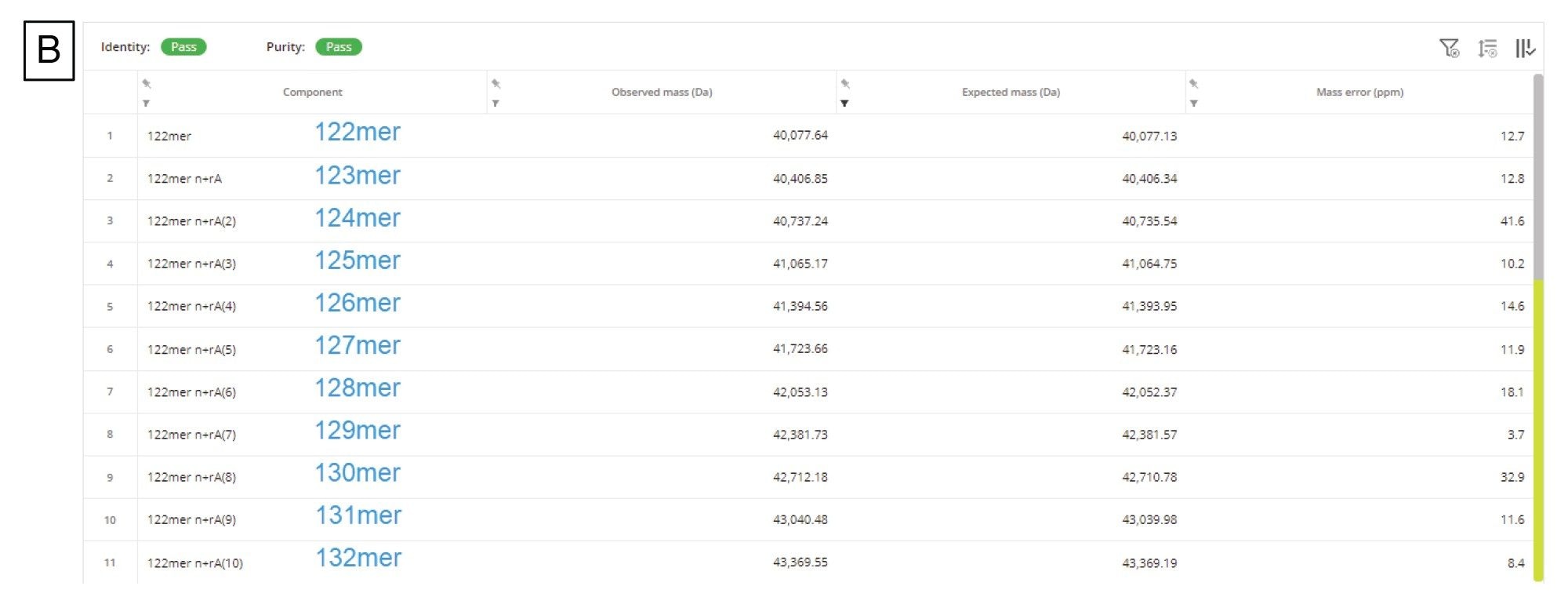 図 10.(B)3' ポリ(A)テールの特性である広い分散度を示す INTACT Mass アプリケーションの結果テーブル。最初に検出されたテールバリアント(122 mer)に最大 10 個のアデノシンの質量が追加されています。カスタムデコンボリューションパラメーターで INTACT Mass 解析を使用して、122 ~ 132 mer の範囲の 11 種類のオリゴヌクレオチドを、45 ppm を上回る質量精度で同定しました。
図 10.(B)3' ポリ(A)テールの特性である広い分散度を示す INTACT Mass アプリケーションの結果テーブル。最初に検出されたテールバリアント(122 mer)に最大 10 個のアデノシンの質量が追加されています。カスタムデコンボリューションパラメーターで INTACT Mass 解析を使用して、122 ~ 132 mer の範囲の 11 種類のオリゴヌクレオチドを、45 ppm を上回る質量精度で同定しました。
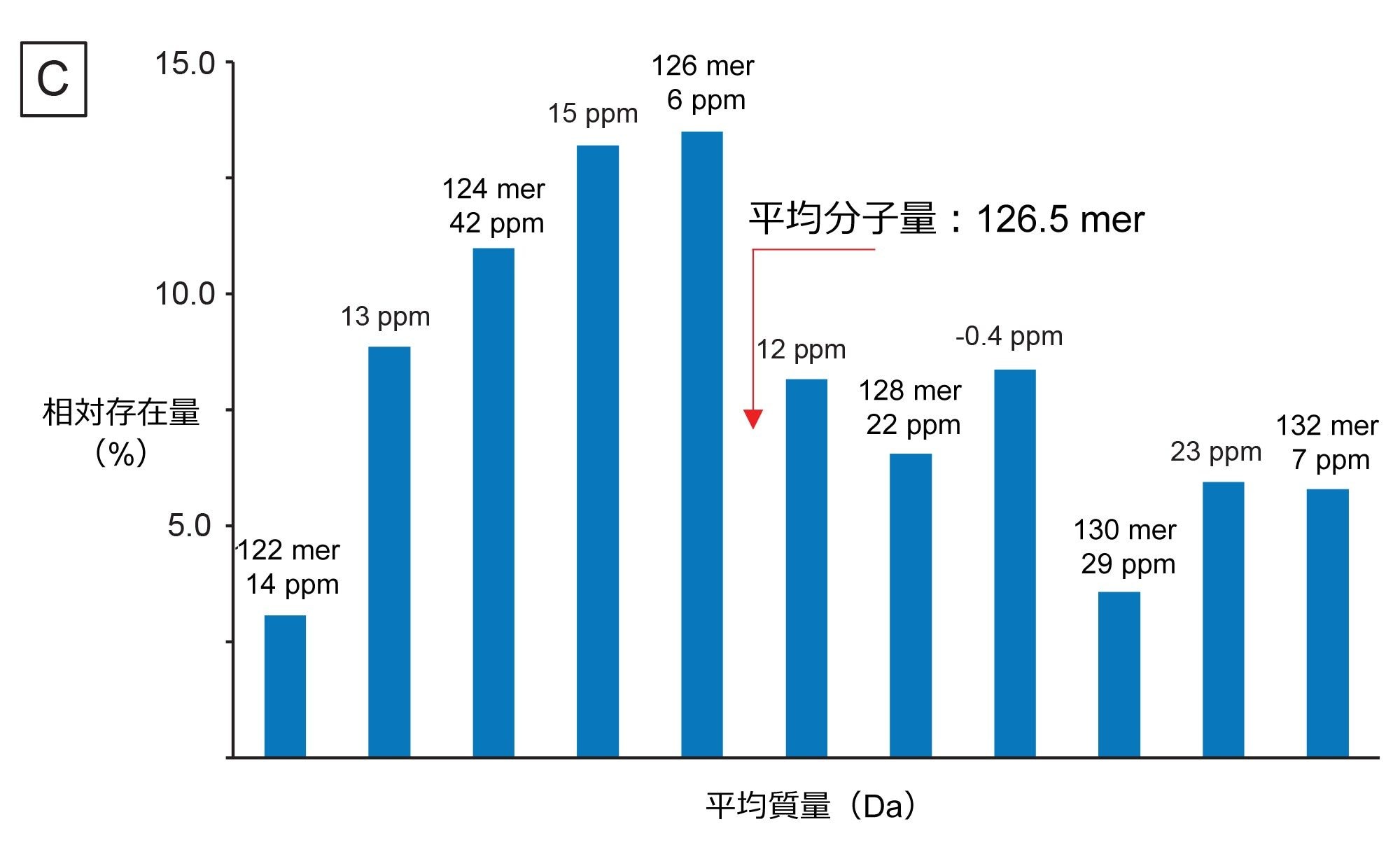 図 10.(C)3' ポリ(A)テールの長さに対する ESI-MS スペクトル強度の分布により、平均テール長と分散度を決定することができます。
図 10.(C)3' ポリ(A)テールの長さに対する ESI-MS スペクトル強度の分布により、平均テール長と分散度を決定することができます。
3' ポリ(A)の分散度と平均質量の測定値はいずれも、医薬品 mRNA 分子において重要な CQA であり、これらによってプロセスの頑健性および潜在的な臨床効果を評価することができます。このように、Doneanu らが開発した LC-MS アッセイおよび INTACT Mass アプリを使用したデータ分析ワークフローは、予防/治療用 mRNA から酵素的に切断された 3' ポリ(A)テールの分析に有効であることがわかります。
結論
- BioAccord LC-MS システムおよび waters_connect インフォマティクスプラットホーム上に構築されたワークフローによって、長いオリゴ医薬品および最新の RNA 医薬品の CQA を、配列のマッピングによる確認(オリゴ、sgRNA、mRNA)、5' キャッピングの有効性(mRNA)、3' ポリ(A)テールの分析(mRNA)などのアプローチで分析することができます。
- これらのワークフローをサポートするために、INTACT Mass アプリケーションのインフォマティクスを機能強化しました。新規のカスタムデコンボリューション機能により、シグナル強度が低くダイナミックレンジが大きいサンプルであっても、対象分析種のルーチンの自動データ解析が可能になります。
- INTACT Mass アプリケーションのこれらの機能強化は、RNA データの分析用に開発された以下の追加のマイクロアプリによって補完されています。
- ユーザーが編集できる残基、酵素、mRNA 修飾が含まれる in silico 消化リストを生成する mRNA Cleaver
- 消化フラグメントマップのシーケンスカバレッジを簡単に視覚化および計算できる Coverage Viewer
参考文献
- Zhu, Y., Zhu, L., Wang, X. & Jin, H. RNA-based Therapeutics: An overview and prospectus.Cell Death & Disease 13, (2022).
- Crooke ST, Witztum JL, Bennett CF, Baker BF.RNA-Targeted Therapeutics.Cell Metab.2018 Apr 3;27(4):714-739.doi: 10.1016/j.cmet.2018.03.004. Erratum in: Cell Metab.2019 Feb 5;29(2):501.PMID: 2961764
- Damase, T. R. et al.The Limitless Future of RNA therapeutics.Frontiers in Bioengineering and Biotechnology 9, (2021).
- Xu, S.; Yang, K.; Li, R.; Zhang, L., mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. International Journal of Molecular Sciences 2020, 21 (18), 6582
- Verbeke, R., Lentacker, I., De Smedt, S. C. &; Dewitte, H. The dawn of mRNA vaccines: The COVID-19 case. Journal of Controlled Release 333, 511–520 (2021).
- Jackson, N. A., Kester, K. E., Casimiro, D., Gurunathan, S. &; DeRosa, F. The Promise of mRNA vaccines: A biotech and industrial perspective.npj Vaccines 5, (2020).
- Goyon, A. et al.Full sequencing of CRISPR/Cas9 single guide RNA (sgRNA) via parallel ribonuclease digestions and hydrophilic interaction liquid chromatography–high-resolution mass spectrometry analysis.Analytical Chemistry 93, 14792–14801 (2021).
- Ivleva, V. B., Yu, Y.-Q. &; Gilar, M. Ultra-performance liquid chromatography/tandem mass spectrometry (UPLC/MS/MS) and UPLC/MSe analysis of RNA oligonucleotides.Rapid Communications in Mass Spectrometry 24, 2631–2640 (2010).
- Jiang, T. et al.Oligonucleotide sequence mapping of large therapeutic mRNAs via parallel ribonuclease digestions and LC-MS/MS.Analytical Chemistry 91, 8500–8506 (2019).
- Ross, R., Cao, X. & Limbach, P. Mapping post‐transcriptional modifications onto transfer ribonucleic acid sequences by liquid chromatography tandem mass spectrometry.Biomolecules 7, 21 (2017).
- Shion, H., Boyce, P., Berger, S. & Yu, Y. Q. INTACT Mass™ - A Versatile waters_connect™ application for Rapid Mass Confirmation and Purity Assessment of Biotherapeutics.Waters Application Note, 720007547, February 2023.
- Jackson, N. A., Kester, K. E., Casimiro, D., Gurunathan, S. & DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective.npj Vaccines 5, (2020).
- Doneanu, C., Fredette, J. & Yu, Y. Q. Ion-Pairing Reversed Phase LC-MS Analysis of Poly(A) Heterogeneity Using the BioAccord LC-MS System, Waters Application Note, 720007925, 2023.
- Nguyen, J. M. et al. Rapid analysis of synthetic mRNA cap structure using ion-pairing RPLC with the BioAccord LC-MS System.Waters Application Note, 720007329, October 2023.
- Gilar, M. Size-exclusion chromatography method for poly(A) tail analysis of mRNA.Waters Application Note, 720007853, October 2023.
- Hu, L., Li, Y., Wang, J., Zhao, Y. & Wang, Y. Controlling CRISPR-Cas9 by guide RNA engineering.WIREs RNA 14, (2022).
- Pelletier, J.; Sonenberg, N. The Organizing Principles of Eukaryotic Ribosome Recruitment.Annu.Rev.Biochem.2019, 88, 307–335
- Gaye, M. M. et al. Synthetic mRNA oligo-mapping using ion-pairing liquid chromatography and mass spectrometry.Waters Application Note, 720007669, October 2023.
- Fekete, S et al.Challenges and emerging trends in liquid chromatography-based analyses of mRNA pharmaceuticals.Journal of Pharmaceutical and Biomedical Analysis.224 (2022).
- Fountain, K. J., Gilar, M. & Gebler, J. C. Analysis of native and chemically modifed oligonucleotides by tandem ion-pair reversed phase high-performance liquid chromatography/electrospray ionization mass spectrometry.Rapid Commun.Mass Spectrom. 17, 646–653. https://doi.org/10.1002/rcm.959 (2003).
- Zhang, G., Lin, J., Srinivasan, K., Kavetskaia, O. & Duncan, J. N. Strategies for bioanalysis of an oligonucleotide class macromolecule from rat plasma using liquid chromatography−tandem mass spectrometry.Anal.Chem.79, 3416–3424.https://doi.org/10.1021/ac0618674 (2007).
- Deng, P., Chen, X., Zhang, G. & Zhong, D. Bioanalysis of an oligonucleotide and its metabolites by liquid chromatography–tandem mass spectrometry.J. Pharm.Biomed.Anal.52, 571–579.https://doi.org/10.1016/j.jpba.2010.01.040 (2010).
- Gau et al.Oligonucleotide mapping via mass spectrometry to enable comprehensive primary structure characterization of an mRNA vaccine against SARS‑CoV‑2.Nature Scientific Reports.13:9038 2023.
- Loverix S, Winqvist A, Stromber R, Steyaert J. Mechanism of RNase T1: concerted triester-like phosphoryl transfer via a catalytic three-centered hydrogen bond Chem Biol.2000 Aug, 7(8):651-8 doi: 10.1016/s1074-5521(00)00005-3.
- Loverix S, Steyaert J. Deciphering the Mechanism of RNase T1.Methods in Enzymology, Academic Press, Vol.341, 2001, 305-323, doi: 10.1016/S0076-6879(01)41160-8
- Masachika Irie.3- RNase T1/RNase T2 Family RNases.Ribonucleases, Academic Press, 1997.101-130, doi: 10.1016/B978-012588945-2/50004-2.
- Sorrentino, Salvatore.The Eight Human “Canonical” Ribonucleases: Molecular Diversity, catalytic properties, and special biological actions of the enzyme proteins.FEBS Letters.Vol 584, Issue 11, 2010, 2194-2200, doi: 10.1016/j.febslet.2010.04.018
- Gaye M.M. et al.CRISPR Single Guide RNA Characterization by IP-RP-LC-MS with a Premier Oligonucleotide BEH 300Å C18 Column.Waters Application Note, 720007897, October 2023.
720008130JA、2023 年 11 月