イオン対液体クロマトグラフィーと質量分析を用いた合成 mRNA のオリゴマッピング
要約
メッセンジャー RNA は、これを用いた癌治療に対する評価および COVID-19 に対する mRNA ワクチンの FDA 承認に示されているように、ヒトの医療にとって急速に重要な治療法になっています。mRNA ワクチンやその他のクラスの mRNA 医薬品の急速な開発は、分析手法の進歩によって支えられています。このような方法の重要な側面の 1 つは、液体クロマトグラフィー質量分析(LC-MS)による配列のマッピングにより、治療用 mRNA のアイデンティティー、純度、修飾を確認することです。RNA の質量分析に基づくシーケンシングは、フラグメントの直接分子検出が提供されるため、テンプレート化による RNA シーケンシングより優れており、これを使用して、ヌクレオチド不純物の位置を特定し、重要な構造特性(5’キャップおよびポリ A テール)を識別することができます。そのため、精密質量マッチから得られる mRNA 成分の注釈付きクロマトグラムをもたらす、mRNA の包括的なボトムアップ LC-UV-MS 特性解析のワークフローを、当社は提案します。
アプリケーションのメリット
- ACQUITY™ Premier Oligonucleotide BEH™ C18 300 Å カラムと組み合わせたイオン対逆相クロマトグラフィーを使用することによる、高いクロマトグラフィー分離能および MS 感度
- in silico mRNA 消化の計算および waters_connect/UNIFI™ サイエンスライブラリーの適用によって促進される精密質量マッチングに基づく、mRNA 消化物の自動注釈
はじめに
SARS-CoV-2 パンデミックは、核酸ベースの医薬品、特に合成 mRNA の急速な開発に刺激を与えました1。 1961 年に Brenner らによって発見されてから 40 年になります2 [i]。mRNA は、癌治療のためのヒトでの臨床試験の開始1–3および米国食品医薬品局による 2 種類の COVID-19 mRNA ワクチンの完全承認(それぞれ 2021 年 8 月および 2022 年 1 月)によって示されているように、大きな可能性がある重要な治療法に進化しています。mRNA ワクチンやその他のクラスの mRNA 医薬品の急速な開発は、分析手法の進歩によって支えられています。このような方法の重要な側面の 1 つは、質量分析と結合した液体クロマトグラフィー(LC-MS)によるオリゴマッピングおよびシーケンシングにより、治療用 mRNA のアイデンティティー、純度、修飾を確認することです。サンガー シーケンシングや次世代シーケンシング(NGS)などの核酸シーケンシングテクノロジーは、医薬品開発者に貴重な情報を提供します。ただし、タンデム MS(LC-MS/MS)4 や MSE(低コリジョンエネルギーと高コリジョンエネルギーを交互に使用)5 ベースのフラグメンテーションと組み合わせて LC を使用することによって達成できる、レベルを上げた分析もあります。プロテオミクスのボトムアップアプローチと同様に、LC-MS/MS や MSE ベースの配列決定では、ヌクレオチド不純物の検出と位置確認、および脂質付加核酸塩基6、末端キャッピングした残基、ポリ A テール修飾のような重要な構造特性など、RNA フラグメントを直接分子検出する利点があります4,7。大量のデータ解析ソリューションが存在するボトムアッププロテオミクスワークフローとは異なり、RNA マッピングの選択肢は限られています。LC、UV 検出、MS 測定で構成される単一プラットホーム内で、所定の合成 mRNA を特性解析するボトムアップアプローチに基づく、オリゴマッピングのワークフローを、当社は提案します。消化物成分を、社内で開発した自由に使用できる in silico 消化ライブラリーカリキュレーター(mRNAcalcondemand)と waters_connect™ を組み合わせて解析し、注釈付きクロマトグラムを生成しました。ここでは、RNase T1 消化ルシフェラーゼ mRNA を使用して、mRNA 配列マッピングの分析アプローチを実証します。
実験方法
サンプル情報
Bijoyita Roy(New England Biolabs、マサチューセッツ州 Ipswich)からご提供いただいた Cypridina ルシフェラーゼ mRNA(キャップなし、ポリ A テール修飾なし)約 90 µg を、3’-グアノシンの特定の RNaseT1(Worthington Biochemical Corporation、ニュージャージー州 Lakewood)を使用して消化しました。このワークフローを、10 µg の TriLink Biotechnologies(CleanCap®FLuc mRNA、カルフォルニア州 San Diego)のホタルルシフェラーゼ mRNA(未翻訳の配列は特許取得済み)を使用して繰り返したところ、同等の結果が得られました。ルシフェラーゼ mRNA は、消化の前に、ヌクレアーゼフリーバッファー(10 mM トリス、0.1 mM EDTA 水溶液(pH 7.5)、Integrated DNA Technologies, Inc、アイオワ州 Coralville)中に調製した 20 µL 尿素(8 M)を用いて、80 ℃ で 5 分間変性しました。次に、ヌクレアーゼフリーバッファーに再懸濁した 24 µg(約 10 kU)の RNase T1(Worthington Biochemical Corporation、ニュージャージー州 Lakewood)を、変性 mRNA に室温で添加し、この混合液を 37 ℃ で 30 分間培養しました。培養終了時に、ヌクレアーゼフリーバッファー(40 µL)を添加してサンプル合計量を 80 µL にしました。最終アリコートは、ポリプロピレン製 300 µL オートサンプラーバイアル(製品番号:186002639)に移しました。得られた消化物は、それ以上の操作を行わずにイオン対逆相クロマトグラフィー(IP-RPLC)で処理し、その後 BioAccord™ RDa™ 検出器を使用してネガティブイオン化モードで MS 検出しました。
LC 条件
|
LC システム: |
ACQUITY UPLC™ Premier BSM システム(BioAccord システムの一部として) |
|
検出器: |
ACQUITY UPLC TUV 検出器 |
|
波長: |
260 nm |
|
カラム: |
ACQUITY Premier Oligonucleotide BEH C18、2.1 × 150 mm、300 Å、1.7 µm(製品番号:186010541) |
|
カラム温度: |
70 ˚C |
|
サンプル温度: |
4 ˚C |
|
注入: |
5 µL |
|
流速: |
0.4 mL/分 |
|
移動相 A |
0.1% N,N-ジイソプロピルエチルアミン(DIPEA)を IP 試薬とし、1% 1,1,1,3,3,3-ヘキサフルオロイソプロパノール(HFIP)を脱イオン水中に調製 |
|
移動相 B |
0.0375% DIPEA、0.075% HFIP 含有 65:35 アセトニトリル:水 |
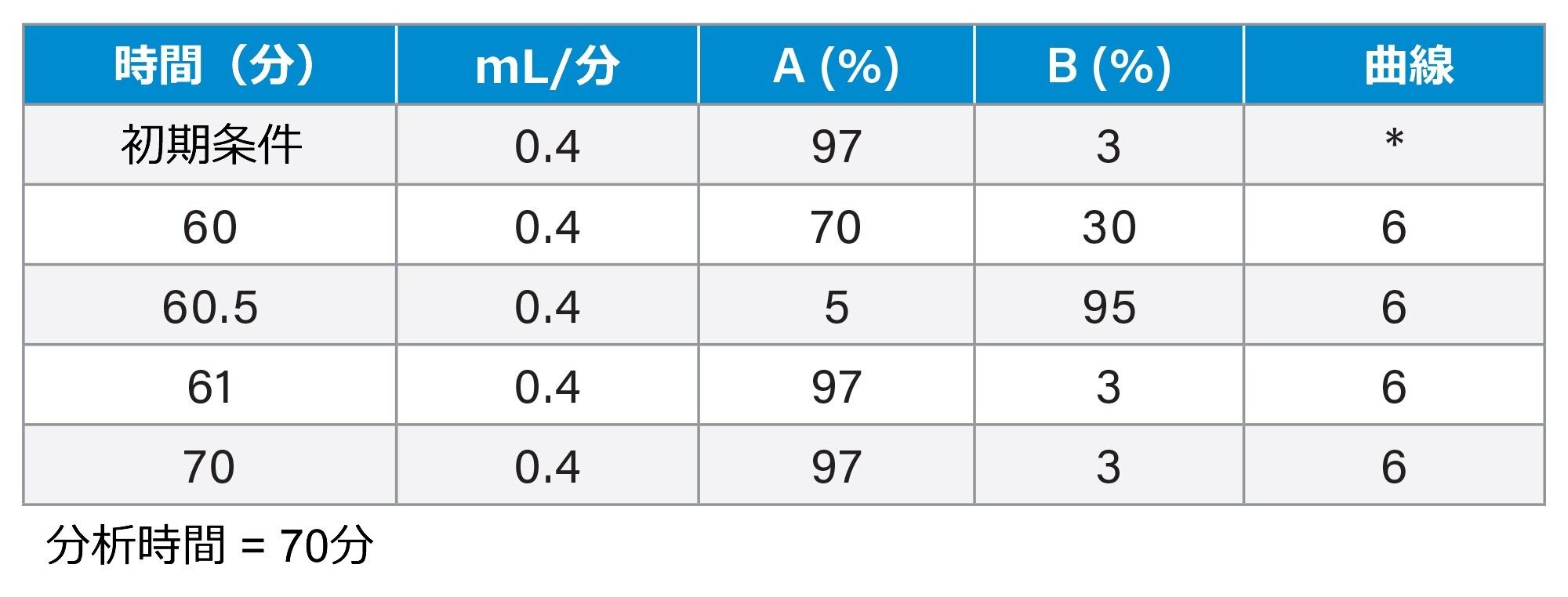
グラジエントテーブル
MS 条件
|
MS システム: |
BioAccord LC-MS システム |
|
検出器: |
ACQUITY RDa 検出器 |
|
モード: |
フラグメンテーションによるフルスキャン |
|
極性: |
ネガティブ |
|
コーン電圧: |
40 V |
|
フラグメンテーションコーン電圧: |
80 ~ 200 V |
|
質量範囲: |
高(m/z 400 ~ 5,000) |
|
スキャンレート: |
2 Hz |
|
キャピラリー電圧: |
0.80 kV |
|
脱溶媒温度: |
400 ℃ |
結果および考察
C18 固定相を用いるイオン対逆相クロマトグラフィー(IP-RPLC)は、オリゴヌクレオチドの分析での実証済みのアプローチになっています4.7。 移動相には、一般にはアルキルアミンなどのイオン対試薬が含まれます。アルキルアミンは C18 固定相 8,9,10 に吸着され、これによって保持機構のようなミックスモードが導入されます8-10。このアプリケーションで使用する N,N-ジイソプロピルエチルアミン(DIPEA)/1,1,1,3,3,3-ヘキサフルオロイソプロパノール(HFIP)移動相システムは、光学 UV 検出およびネガティブイオン化モード質量分析のいずれにも適合します4,7,–10。HFIP は、エレクトロスプレーイオン化を促進するために使用します8。
RNase T1 消化ルシフェラーゼ mRNA を、ACQUITY Premier Oligonucleotide BEH C18(2.1 × 150 mm、300 Å、1.7 µm)カラムに注入し、ACQUITY UPLC TUV 検出器を装着した ACQUITY Premier バイナリー LC を使用してグラジエントを開発しました。この試験で使用した ACQUITY Premier Oligonucleotide BEH C18 カラムは、ACQUITY Premier Oligonucleotide BEH C18 130 Å カラムと類似していますが、ポアサイズが大きいため、より長いオリゴヌクレオチド分子種の分離が向上しています。BioAccord ベンチトップ LC-MS システムの ACQUITY RDa 検出器で、ネガティブイオン化モード質量分析を使用してデータを 3 回繰り返し取り込みました。さらに、後で LC ピーク同定の確認に使用できる高エネルギーフラグメントイオンスペクトルがスキャン 1 回おきに生成されるように、ACQUITY RDa 検出器による MSE データ測定をプログラムしました。図 1 に、RNase T1 対照サンプル(上のトレース)、mRNA 対照サンプル(下のトレース)、消化済み mRNA(中央のトレース)のトータルイオンクロマトグラム(TIC)が、示されています。RNase T1 によるルシフェラーゼ mRNA の消化で生じたオリゴヌクレオチドフラグメントは、4 シグマピークキャパシティ 613 で容易に分離されました。全体として、クロマトグラフィーピークはシャープかつ対称で、3 回繰り返し注入にわたる保持時間(RT)のばらつきは約 0.01 分でした。グラジエント時間 60 分間のメソッドで、消化物の成分は 2 ~ 23 分で溶出し、消化が不完全な一部の mRNA は 29 分付近、インタクト RNaseT1 は 54 分付近で溶出しました。図 1 の上のトレースに示されているように、RNase T1 対照サンプルでは、保持時間 50 分の後にのみシグナルが見られました。これにより、RNase T1 によって mRNA 消化済み成分の保持時間ウィンドウ内に干渉が持ち込まれないことが確認されました(図 1、中央のトレース)。同様に、図 1 の下のトレースにより、インタクトルシフェラーゼ mRNA は約 38 分に溶出することが示されており、消化済みサンプルで 29 分に観察されたピーク(図 1、中央のトレース)は、消化が不完全な mRNA に対応することが確認されます。ここで、5’キャップ構造とポリ A テール構造で構成される合成 mRNA の場合、29 分と 37 分付近のピークが消化後に観察されることがわかります。約 38 分から約 37 分へのわずかなシフトは、消化されなかったポリ A 構造を示す可能性があります(このクロマトグラフィーの動作の検討については、今後のアプリケーションノートで説明します)。
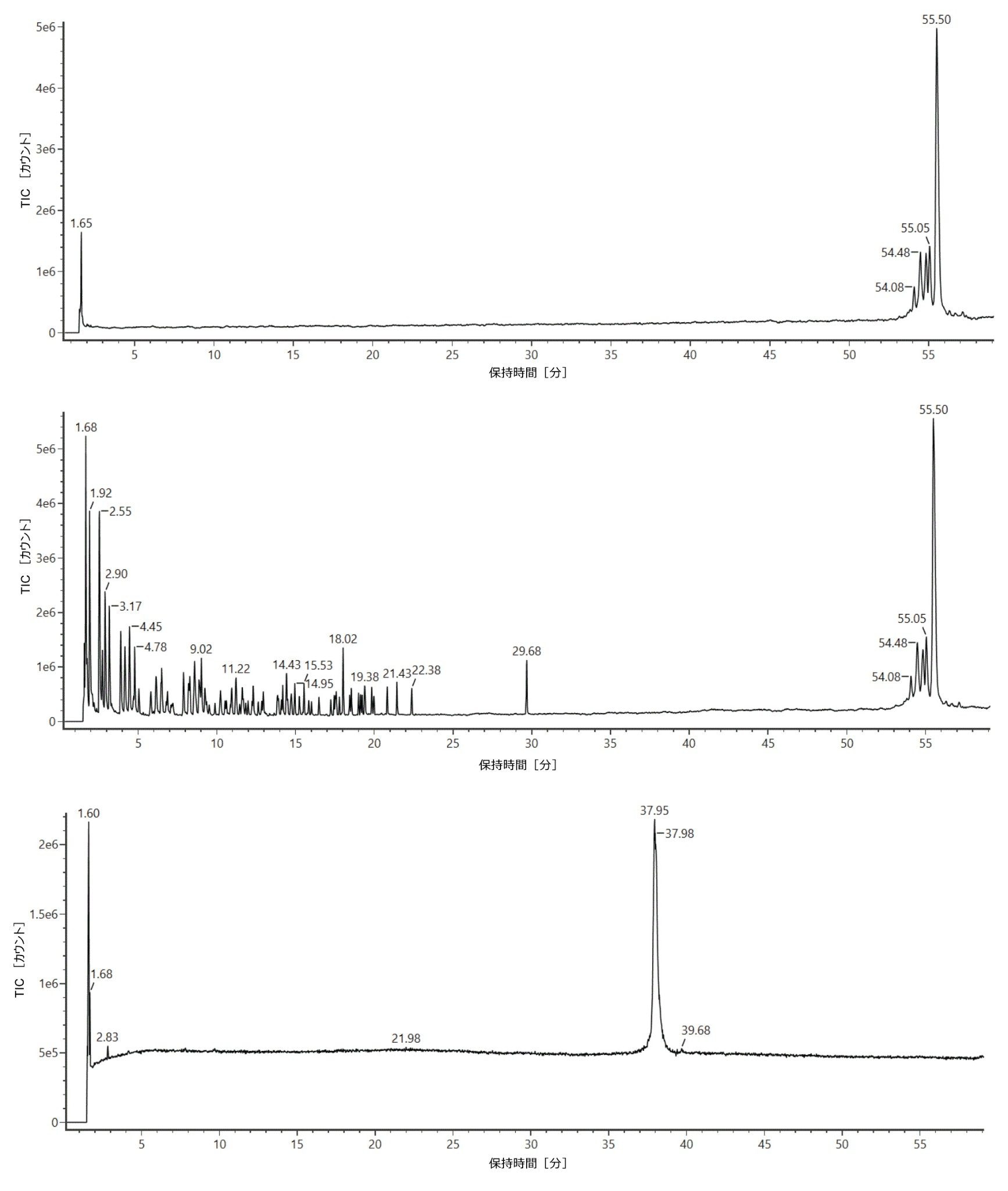 図 1:RNase T1 対照サンプル(上のトレース)、mRNA 対照サンプル(下のトレース)、および RNase T1 で消化し、ACQUITY UPLC I-Class システム(ACQUITY Premier Oligonucleotide BEH C18 カラム、2.1 x 150 mm、300 Å、1.7 µm)およびネガティブイオン化モードの BioAccord ACQUITY RDa 検出器を使用して分析したルシフェラーゼ mRNA のイオン対逆相クロマトグラフィー(IP-RPLC)で得られた消化物(中間トレース)の TIC クロマトグラム。
図 1:RNase T1 対照サンプル(上のトレース)、mRNA 対照サンプル(下のトレース)、および RNase T1 で消化し、ACQUITY UPLC I-Class システム(ACQUITY Premier Oligonucleotide BEH C18 カラム、2.1 x 150 mm、300 Å、1.7 µm)およびネガティブイオン化モードの BioAccord ACQUITY RDa 検出器を使用して分析したルシフェラーゼ mRNA のイオン対逆相クロマトグラフィー(IP-RPLC)で得られた消化物(中間トレース)の TIC クロマトグラム。
in-silico 消化 mRNA カリキュレーター(mRNAcalcondemand)のグラフィカルユーザーインタフェース(GUI)がスキーム 1 に示されています。塩基配列の隣に、修飾、酵素、欠落している開裂など、いくつかの消化パラメーターが指定されています。カリキュレーターにより、この情報に基づいてチャージ状態や m/z 範囲などの MS 固有の設定、およびモノアイソトピック質量または平均質量に基づいて計算を行う機能が、既定に設定されます。生成された出力は、フラットテキスト csv ファイル形式であり、UNIFI または waters_connect ソフトウェアで活用、または補完するダウンストリーム分析に使用できます。
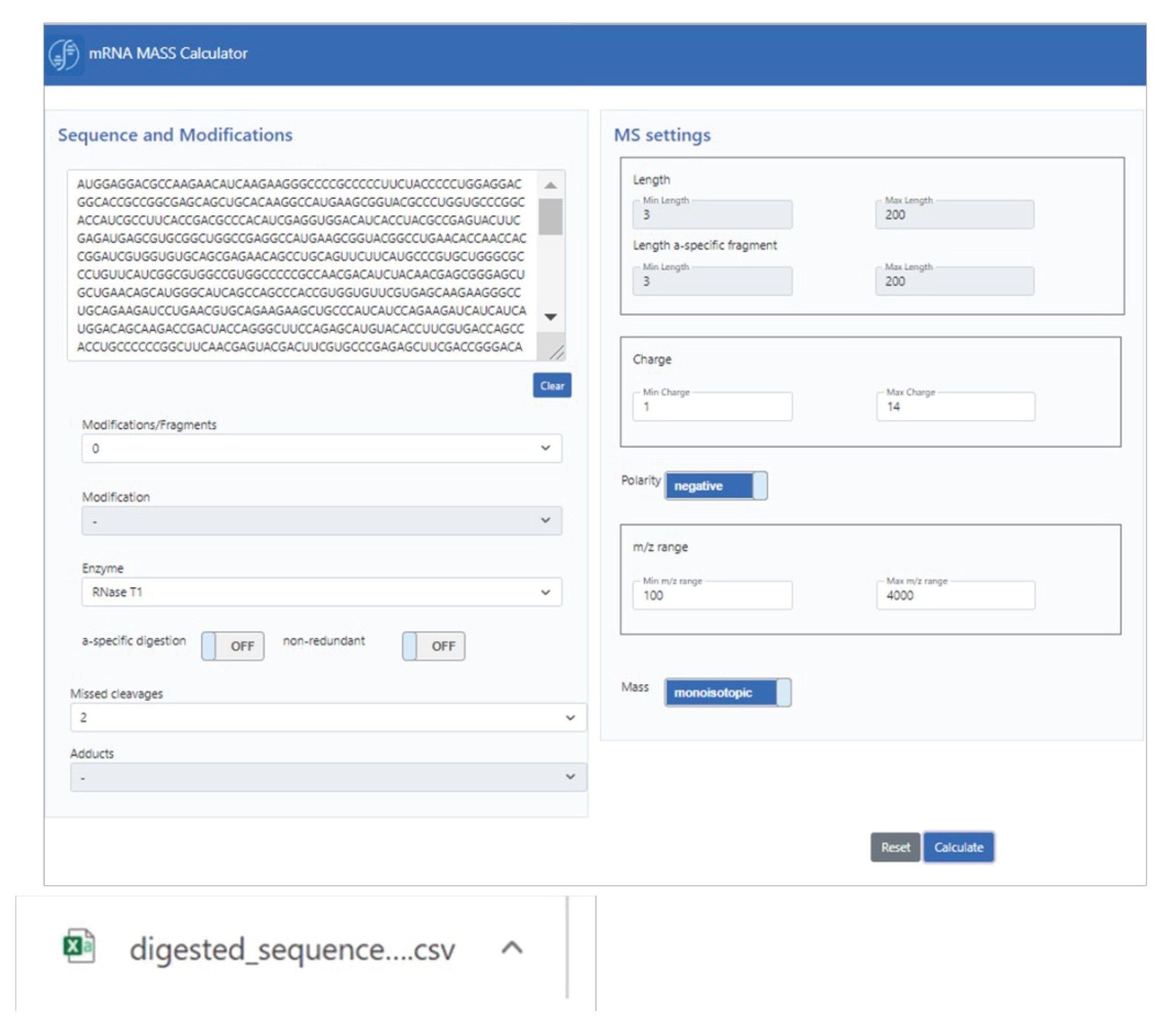 スキーム 1:in silico mRNA 消化物質量計算用の mRNA カリキュレーターの GUI。
スキーム 1:in silico mRNA 消化物質量計算用の mRNA カリキュレーターの GUI。
次に、ライブラリーを作成し、ここでは消化物成分は個々の分析種と見なされます。これは、カリキュレーターの出力をスプレッドシートとして UNIFI サイエンスライブラリーにインポートすることで達成されました。作成したライブラリーを使用して、HRMS スクリーニング分析メソッドで、取り込み後の消化物化合物をターゲットとし、ユーザー指定の許容範囲を用いることができます。質量許容範囲ベースのライブラリーマッチの使用が、以下に示されています。ライブラリー検索の結果、注釈付きクロマトグラムが自動的に生成されます(図 2)。保持時間ウィンドウ 17 ~ 20 分の拡大表示に対応するクローズアップが、図 2 に示されています。
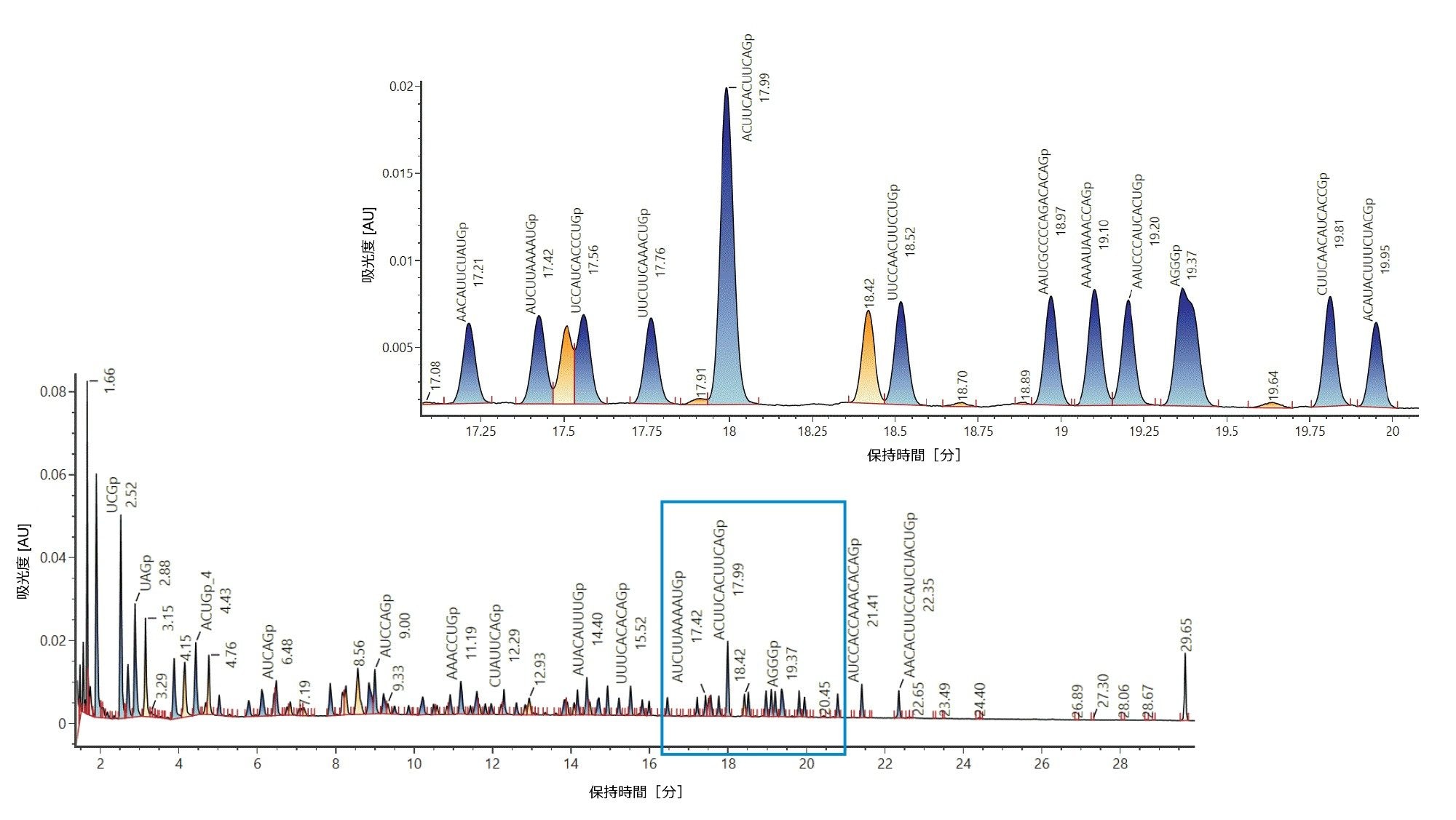 図 2:精密質量に基づいて、ターゲット成分ライブラリーにマッチした後に生成された、ルシフェラーゼ mRNA 消化物の注釈付き TIC クロマトグラム。ルシフェラーゼ mRNA を、RNase T1 で消化し、ACQUITY Premier BSM LC(ACQUITY Premier Oligonucleotide BEH C18 カラム、2.1 × 150 mm、300 Å、1.7 µm)およびネガティブモードの BioAccord ACQUITY RDa 検出器を使用して分析しました。ターゲット成分は mRNA 質量カリキュレーターを使用して計算しました。
図 2:精密質量に基づいて、ターゲット成分ライブラリーにマッチした後に生成された、ルシフェラーゼ mRNA 消化物の注釈付き TIC クロマトグラム。ルシフェラーゼ mRNA を、RNase T1 で消化し、ACQUITY Premier BSM LC(ACQUITY Premier Oligonucleotide BEH C18 カラム、2.1 × 150 mm、300 Å、1.7 µm)およびネガティブモードの BioAccord ACQUITY RDa 検出器を使用して分析しました。ターゲット成分は mRNA 質量カリキュレーターを使用して計算しました。
最大 2 つの開裂の欠落を許容して作成した in silico ライブラリーに対するデータスクリーニングの各技術的繰り返しから、計 436、428、441 の消化成分の可能性のある同定(ID)が生成されました。手動バリデーションのために、いくつかの基準を検討しました。同定された成分 441 のうち 40 は、存在量およびピーク形状(低存在量のクロマトグラフィーピークのショルダーなど)に基づいて除外しました。これらの除外した ID の大部分(40 のうち 27)は、24 ~ 60 分に位置していました。全体として、同定およびバリデーションした成分の約 60% が、質量誤差 10 ppm の範囲内でした(261 消化物成分)。RNase T1 対照サンプル(図 1、上のトレース)に同じクエリーを行いましたが、予想したとおり同定は得られませんでした。ピーク形状と存在量に加えて、同位体分布の信頼性の高い解明に基づいて、結果をさらにバリデーションし、電荷割り当てを推定して、139 の消化物成分(質量誤差 5 ppm 以内が 65%)が得られました。最後に、一部の割り当てを分析に含めませんでした。その理由は、クロマトグラフィーピークの広がりやショルダーピークの繰り返し検出によってトリガーされる、余剰の割り当てと思われたためです。これにより、同定数がさらに 16 成分減少しました。ただし、2 つの例で、分析種の質量が複数の保持時間で現れ、2 つの明瞭な明確に定義されたクロマトグラフィーピークに対応する一意の 2 つの ID が生成されました。異性体 ID(化学組成は同じで配列が異なる分子種)と同重体 ID(化学組成が異なるが [精密質量が異なる] ノミナル質量が似ている分子種)が存在することにも気付きました。最終的に、精密質量マッチに基づいて 90 の一意の成分を同定でき、これらは、異性体のヌクレオチド配列および同重体イオン(表 1 でグレーで強調表示されているセル)を含めて、表 1 に報告されています。
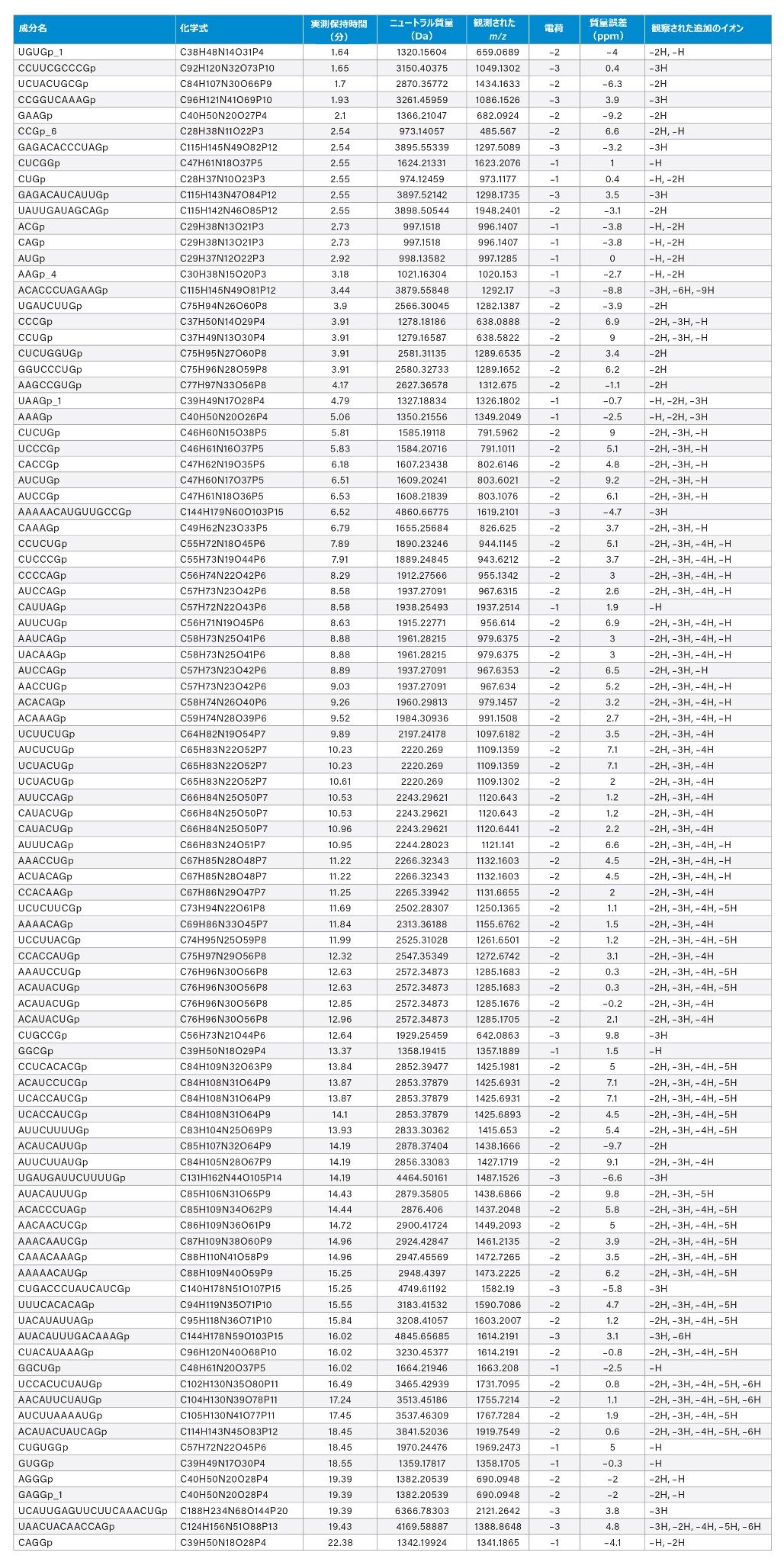 表1:精密質量マッチに基づいて、10 ppm の質量誤差範囲内で暫定的に同定およびバリデーションした、ルシフェラーゼ mRNA 消化物成分。異性体配列または同重体イオンに対応するセルは、グレーで強調表示されています。
表1:精密質量マッチに基づいて、10 ppm の質量誤差範囲内で暫定的に同定およびバリデーションした、ルシフェラーゼ mRNA 消化物成分。異性体配列または同重体イオンに対応するセルは、グレーで強調表示されています。
図 3 に、消化成分 UCCACUCUAUGp の同定が得られたデータ例が示されています。この成分は、左上のクロマトグラムに表示されているように 16.49 分に溶出し、in silico 消化ライブラリーで、m/z 576.5732([M-6H]6-)、692.0876([M-5H]5-)、865.3603([M-4H]4-)、1154.1438([M-3H]3-)、1731.7095([M-2H]2-)の、2 ~ 6 個の負電荷がある 5 つのイオン(図 3、左下のトレース)に基づいて同定されました。[M-5H]5- イオンおよび [M-2H]2- イオンの同位体分布が示され、チャージ状態の割り当てを裏付けるデータが示されています(図 3、右)。
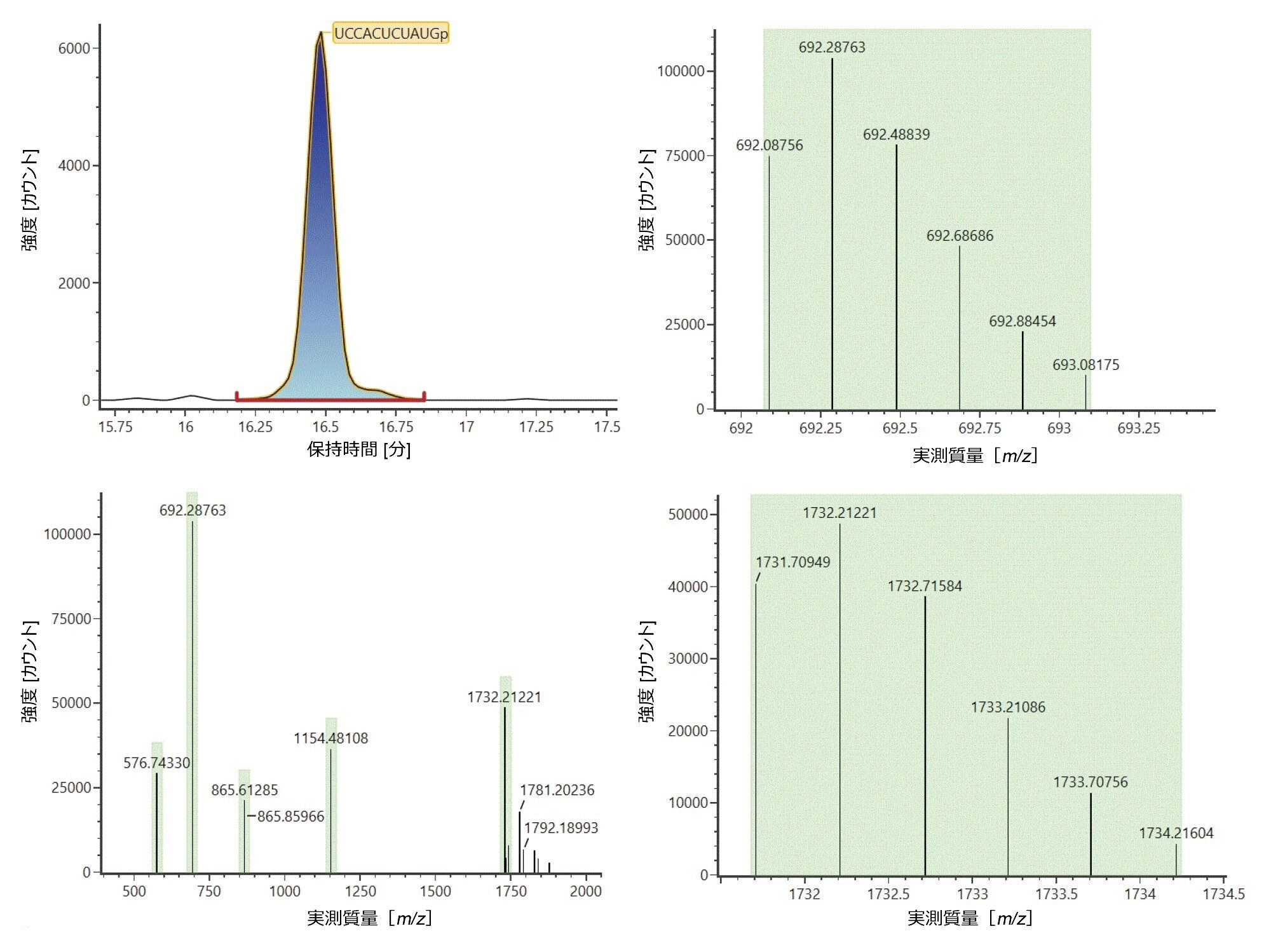 図 3:16.49 分に溶出する消化物成分 UCCACUCUAUGp の同定(左上のトレース)。この成分は、m/z 576.5732([M-6H]6-)、692.0876([M-5H]5-)、865.3603([M-4H]4-)、1154.1438([M-3H]3-)、1731.7095([M-2H]2-)の、2 ~ 6 個の負電荷を帯びた 5 つのイオンに基づいて生成された in silico 消化物から、同定されました。実験的に観察された [M-5H]5- イオンと [M-2H]2- イオンの同位体分布が、右上のトレースと下のトレースにそれぞれ示されています。
図 3:16.49 分に溶出する消化物成分 UCCACUCUAUGp の同定(左上のトレース)。この成分は、m/z 576.5732([M-6H]6-)、692.0876([M-5H]5-)、865.3603([M-4H]4-)、1154.1438([M-3H]3-)、1731.7095([M-2H]2-)の、2 ~ 6 個の負電荷を帯びた 5 つのイオンに基づいて生成された in silico 消化物から、同定されました。実験的に観察された [M-5H]5- イオンと [M-2H]2- イオンの同位体分布が、右上のトレースと下のトレースにそれぞれ示されています。
異性体または同重体イオンの存在による曖昧さは、表 2 に報告されているように、waters_connect CONFIRM Sequence™ アプリケーションを使用して MSE スペクトルを解釈することで解決されました。位置 623 ~ 631(ACAUCCUCGp)および 551~559(UCACCAUCGp)の異性体配列は、RNase T1 の in silico 消化物から予測され、どちらも同じ保持時間 13.87 分に割り当てられています。インタクト質量分析を使用して正しい割り当てを行うことはできません。同じ注入で得られた MSE データを用い、waters_connect CONFIRM Sequence™ アプリケーションを使用してこの割り当ての正しい配列を解析しました。そこでは、各配列に対して高エネルギーフラグメントイオンを予測し、カスタマイズされたアルゴリズムで波形解析した生データの同位体クラスターにマッチしました。確認されたフラグメントイオンをドットマップ上に示すことで、シーケンスカバー率を速やかに評価できます(図 4、B)。UCACCAUCGp について、シーケンス完全カバー率が得られ、正しい割り当てとして容易にバリデーションできました(表 2、インデックス番号 63)。
手動でバリデーションした消化成分(表 1)について得られた中性プリカーサー質量は、973.1406 Da(CCGp、RT 2.54 分)から 6366.7830 Da(UCAUUGAGUUCUUCAAACUGp、RT 19.39 分)の 20-mer ヌクレオチドまでにわたっていました。最も早く溶出した成分は UGUGp で、実測中性質量 1320.1560、保持時間 1.64 分と特定されました。手動でバリデーションした一連の ID の中で溶出する最後のルシフェラーゼ mRNA 消化物成分(表 1)は CAGGp(1342.1992 Da)で、22.38 分に溶出するのが観察されました。このような遅い保持時間での CAGGp の溶出は、密接に関連する配列 UAAGp が約 4 分に溶出したことを考えて、予想外でした。疑陽性の問題に対処するため、waters_connect CONFIRM Sequence アプリケーションを使用して、成分の ID をさらに特性解析しました。MSE データが含まれている MS データに基づいてバリデーションしました。表 2 に示されているように、90 成分のうち CAGGp を含む 34 成分では、精密質量マッチによって配列割り当てがサポートされましたが、配列割り当てをそれ以上バリデーションするために十分な数の MSE フラグメントが生成されませんでした。もう 1 つの興味深い別の例は、割り当てられた配列 AAAAACAUGUUGCCGp(4860.6678 Da、15-mer、プリン 9 個)と AUACAUUUGACAAAGp(4845.6568 Da、15-mer、プリン 9 個)の同じデータセット内で、同じプリン含有量で 10 分離れた溶出が観察されたことです。MSE データを使用して、AAAAACAUGUUGCCGp を疑陽性として除外し、AUACAUUUGACAAAGp の同定を確認することができました。これにより、精密質量マッチとフラグメンテーションスペクトルの両方を戦略的に用いて、消化済み mRNA 配列から得られた成分を曖昧さなしに同定する補助とすることの、重要性が実証されています。さらに、得られた結果から、消化済み mRNA 成分の保持は、最初に考えたほど予測可能ではない可能性があること、およびオリゴヌクレオチドとクロマトグラフィー固定相の相互作用をさらに詳しく調べてモデル化することが急務であることを認識しました。
最後に、手動でバリデーションしたマッチ済み消化物成分を mRNA 配列と比較することで、シーケンスカバー率を手動で推定しました。401 の初期のマッチのシーケンスカバー率の暫定推定で、カバー率の値約 76% が得られました。厳密にバリデーションしたマッチを mRNA 配列に対して確認したところ、得られたカバー率の値は約 22% でした。観察された消化成分の多くはルシフェラーゼ mRNA 配列の複数の位置にマッピングされ(表 2)、修飾または完全修飾された核酸配列にはわずか 4 種類の一意の残基のみが存在するため、冗長性が期待されました。
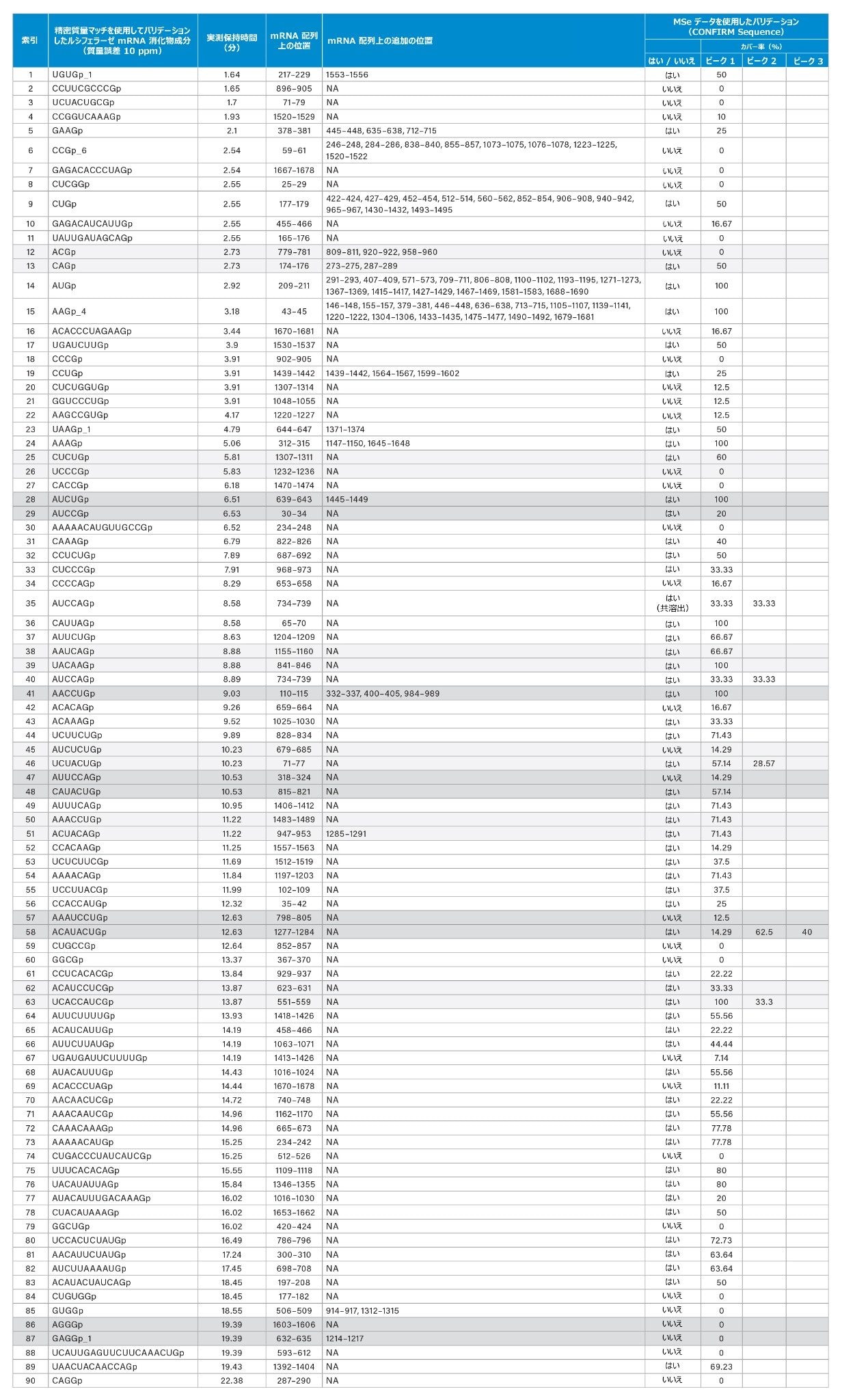 表 2: 精密質量マッチ、および waters_connect CONFIRM Sequence アプリケーションと収集した MSE スペクトルを使用したさらなるバリデーションに基づく、ルシフェラーゼ mRNA 消化成分の同定およびバリデーション。
表 2: 精密質量マッチ、および waters_connect CONFIRM Sequence アプリケーションと収集した MSE スペクトルを使用したさらなるバリデーションに基づく、ルシフェラーゼ mRNA 消化成分の同定およびバリデーション。
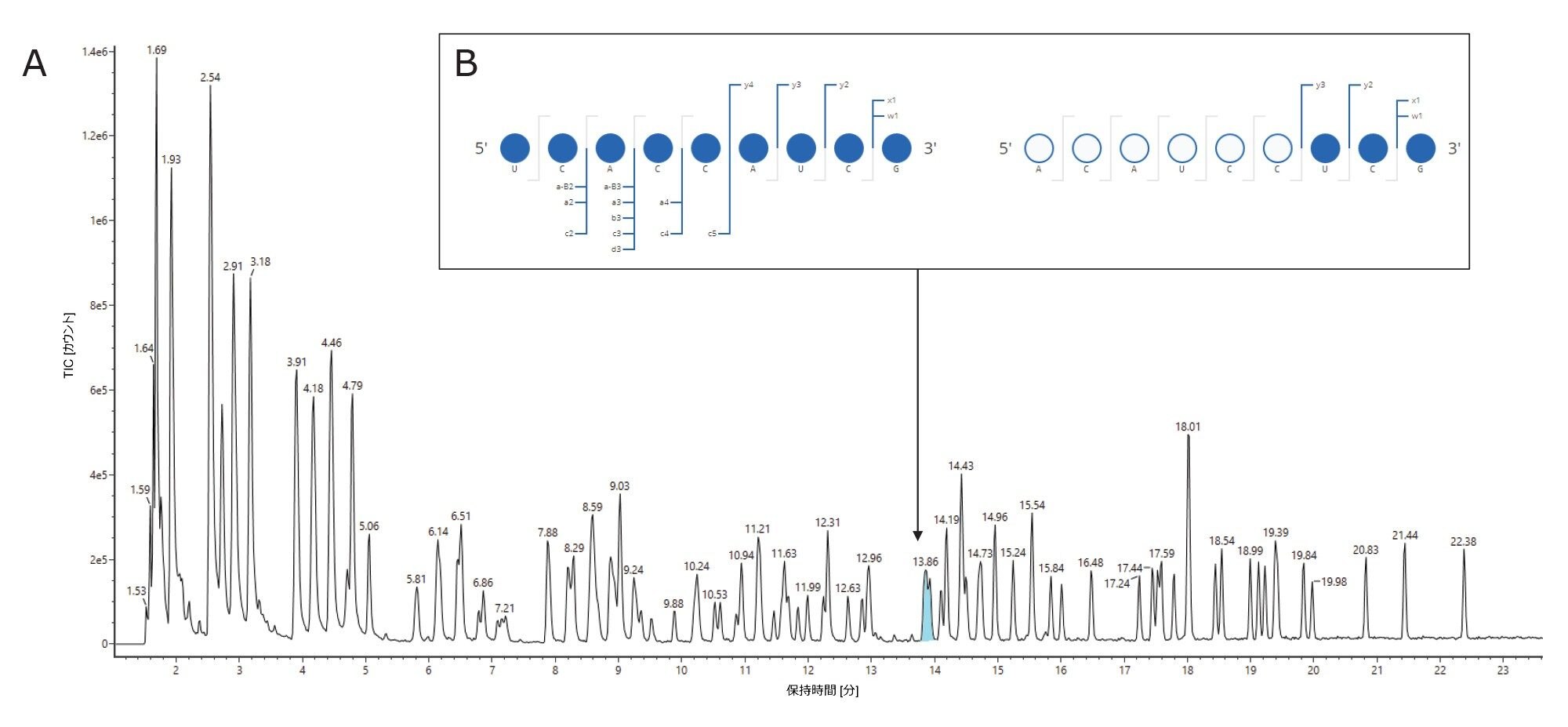 図 4:(A) 位置 623 ~ 631(ACAUCCUCGp)および 551 ~ 559(UCACCAUCGp)の消化済みフラグメント成分が RNase T1 消化物から予測され、TIC の同じ RT のピークに割り当てられます。インタクト質量情報を使用して正しい割り当てを決定することはできません。(B)同じ注入から得られた MSE データを使用して、この割り当ての正しい配列を解明できます。waters_connect CONFIRM Sequence アプリケーションを用い、McLucky の注釈11を使用して各配列に対して高エネルギーフラグメントイオンを予測し、カスタマイズされたアルゴリズムで波形解析した生データの同位体クラスターにマッチしました。このソフトウェアにより、確認済みのフラグメントイオンがドットマップに提示され、シーケンスカバー率がすばやく評価されます。
図 4:(A) 位置 623 ~ 631(ACAUCCUCGp)および 551 ~ 559(UCACCAUCGp)の消化済みフラグメント成分が RNase T1 消化物から予測され、TIC の同じ RT のピークに割り当てられます。インタクト質量情報を使用して正しい割り当てを決定することはできません。(B)同じ注入から得られた MSE データを使用して、この割り当ての正しい配列を解明できます。waters_connect CONFIRM Sequence アプリケーションを用い、McLucky の注釈11を使用して各配列に対して高エネルギーフラグメントイオンを予測し、カスタマイズされたアルゴリズムで波形解析した生データの同位体クラスターにマッチしました。このソフトウェアにより、確認済みのフラグメントイオンがドットマップに提示され、シーケンスカバー率がすばやく評価されます。
結論
本研究で、IP-RPLC と MS を用いた、合成 mRNA のオリゴマッピング用の頑健な分析ワークフローを確立しました。
- 合成 mRNA を、わずか 10 µg の試料から出発して、RNase T1 を用いて高い再現性で消化し、追加のサンプルクリーンアップなしで ACQUITY Premier Oligonucleotide BEH C18(2.1 x 150 mm、300 Å、1.7 µm)カラムに注入しました
- ACQUITY Premier LC でイオン対逆相クロマトグラフィーを使用して、高いクロマトグラフィー分離が達成され、これにより、消化物成分を消化が不完全な mRNA および残存する酵素から容易に分離でき、BioAccord ACQUITY RDa 検出器で効率的に検出できました
- in silico での mRNA 消化計算および waters_connect/UNIFI サイエンスライブラリーの適用によって促進される精密質量マッチに基づいて、注釈付き mRNA 消化クロマトグラムを生成しました
- 消化済み成分に割り当てた配列は、waters_connect CONFIRM Sequence アプリケーションを用いて、MSE スペクトルに基づいてさらにバリデーションしました。さらに、ドットマップによる視覚化を使用して、可能性のある割り当てのフラグメントイオンカバー率を速やかに確認しました
本研究の目的は、mRNA 分子のボトムアップ特性解析を容易にするために必要な、クロマトグラフィー、検出、データ解釈のアプローチを確立することでした。RNase T1 消化は、データの収集と分析のワークフローを確立する、最初の例および概念の証明としてのみ、適用しました。とはいえ、(1)複数のさまざまなヌクレアーゼを用いて補完性のある追加の配列マッピング情報を生成する、および(2)データ取り込みに多重化アプローチを採用することにより、所定の mRNA 構造をより包括的に精査する機会は十分にあります。包括的なシーケンスカバー率の達成を目的とするこれらの側面については、今後の研究で詳しく探査されるでしょう。
参考文献
- Xu, S.; Yang, K.; Li, R.; Zhang, L., mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection.International Journal of Molecular Sciences 2020, 21 (18), 6582.
- Brenner, S.; Jacob, F.; Meselson, M., An Unstable Intermediate Carrying Information From Genes to Ribosomes for Protein Synthesis. Nature 1961, 190 (4776), 576–581.
- Weide, B.; Pascolo, S.; Scheel, B. Derhovanessian, E.; Pflugfelder, A.; Eigentler, T. K. Pawelec, G.; Hoerr, I.; Rammensee, H. G.; Garbe, C., Direct Injection of Protamine-Protected Mrna: Results of a Phase 1/2 Vaccination Trial in Metastatic Melanoma Patients.J Immunother 2009, 32 (5), 498–507.
- Jiang, T.; Yu, N.; Kim, J.; Murgo, J.-R.; Kissai, M.; Ravichandran, K.; Miracco, E. J.; Presnyak, V.; Hua, S., Oligonucleotide Sequence Mapping of Large Therapeutic mRNA s via Parallel Ribonuclease Digestions and LC-MS/MS.Anal.Chem.2019, 91 (13), 8500–8506.
- Plumb, R. S.; Johnson, K. A.; Rainville, P.; Smith, B. W.; Wilson, I. D.; Castro-Perez, J. M.; Nicholson, J. K., UPLC/MSE; A New Approach for Generating Molecular Fragment Information for Biomarker Structure Elucidation.Rapid Communications in Mass Spectrometry 2006, 20 (13), 1989–1994.
- Packer, M.; Gyawali, D.; Yerabolu, R.; Schariter, J.; White, P., A Novel Mechanism for the Loss of mRNA Activity in Lipid Nanoparticle Delivery Systems.Nat.Commun.2021, 12 (1), 6777.
- Goyon, A.; Scott, B.; Kurita, K.; Maschinot, C.; Meyer, K.; Yehl, P.; Zhang, K., On-line Sequencing of CRISPR Guide RNAs and Their Impurities via the Use of Immobilized Ribonuclease Cartridges Attached to a 2D/3D-LC–MS System.Anal.Chem.2021.
- Guo, L.; Worth, A. J.; Mesaros, C.; Snyder, N. W.; Glickson, J. D.; Blair, I. A., Diisopropylethylamine/Hexafluoroisopropanol-Mediated Ion-Pairing Ultra-High-Performance Liquid Chromatography/Mass Spectrometry for Phosphate and Carboxylate Metabolite Analysis: Utility for Studying Cellular Metabolism.Rapid Commun Mass Spectrom 2016, 30 (16), 1835–1845.
- Birdsall, R. E.; Gilar, M.; Shion, H.; Yu, Y. Q.; Chen, W., Reduction of Metal Adducts in Oligonucleotide Mass Spectra in Ion-Pair Reversed-Phase Chromatography/Mass Spectrometry Analysis.Rapid Commun Mass Spectrom 2016, 30 (14), 1667–1679.
- Fountain, K.; Gilar, M.; Budman, Y.; Gebler, J., Purification of Dye-Labeled Oligonucleotides by Ion-Pair Reversed-Phase High-Performance Liquid Chromatography.Journal of chromatography.B, Analytical technologies in the biomedical and life sciences 2003, 783, 61–72.
- McLuckey, S. A.; Van Berkel, G. J.; Glish, G. L., Tandem Mass Spectrometry of Small, Multiply Charged Oligonucleotides.J. Am.Soc.Mass Spectrom.1992, 3 (1), 60–70.
謝辞
本研究に貴重な貢献をしたウォーターズの同僚 Ana-Maria Rotaru、Emanuela Petreanu、Claudia Florea、Dave Jackson、Simon Jones に感謝致します。New England Biolabs での共同研究者である Bijoyita Roy、Siuhong Chan、Ivan R. Corrêa Jr.、Erbay Yigit、G. Brett Robb に、ルシフェラーゼ mRNA および多くの議論を提供して頂いたことについて、感謝致します。
720007669JA、2022 年 6 月