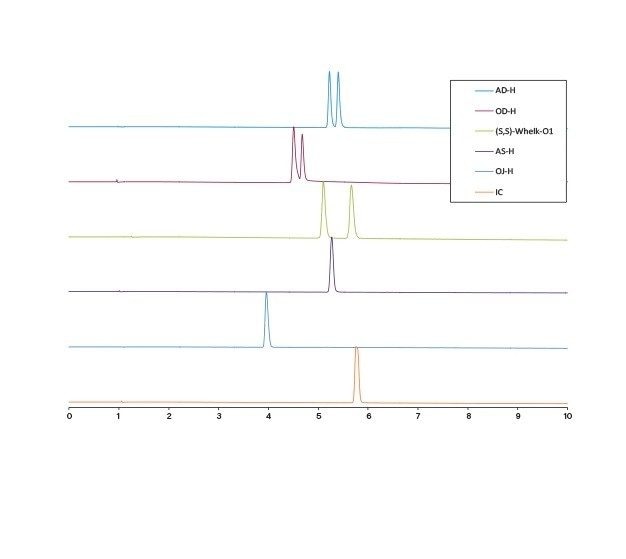
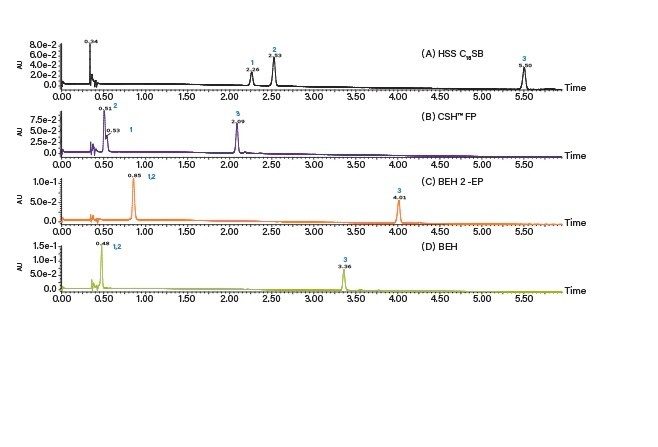
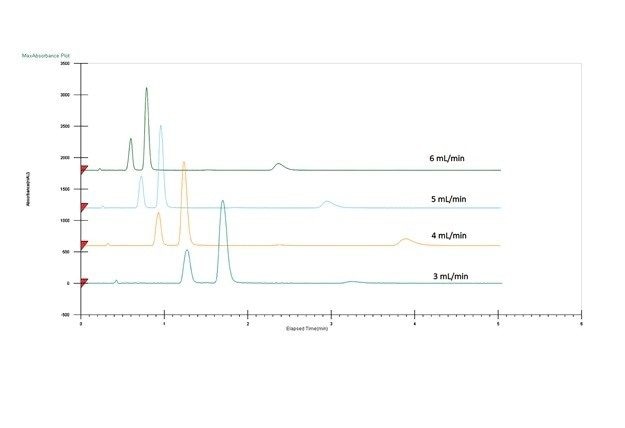
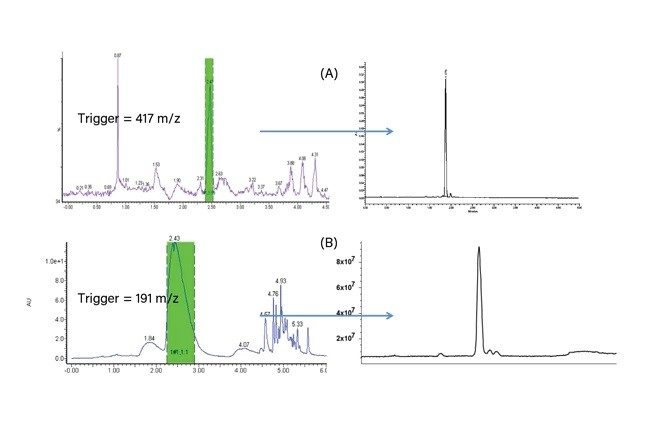
移動相条件の最適化
溶媒とカラムの最適な組み合わせの選択は、アプリケーションまたは目的により異なります。完全なシナリオとしては、高い負荷量、良好な分離能および迅速な分離を組み合わせることで、ターゲット化合物を高純度で完全に回収することができます。しかしながら現実には、最も生産性の高いソリューションを決定するために取捨選択が必要になります。例えば、ピークは良好に分離できても(高い負荷量が可能)、より長い実行時間が必要になる場合があります。一方、分離が良好で、実行時間が非常に短い場合、サイクル時間が短縮しますが、負荷量が低下する可能性があります。別の検討事項としてはフラクションの分取、その後の処理があり、溶媒の量や種類が重要な要素となる可能性があります。最後に、分離をアイソクラティック条件で実施できる場合には、スタックインジェクションの利用が可能で、生産性が大幅に向上します。カラムと溶媒を選択したら、スケールアップおよび精製の前にその他のパラメーターを操作して分離をさらに最適化できます。
グラジエント条件の最適化:SFC では、フォーカスグラジエントは RPLC と同じようには使用できません。順相クロマトグラフィーでは競合する保持メカニズムが多数あるため、グラジエントの変化は各分析種の保持に対して全く異なる影響を与えることになります。SFC では特に、グラジエントにおける共溶媒の増加に伴う密度変化および圧力降下もあります。共溶媒の量が増えると、その保持時間に対する影響は直線的ではないため、変化する条件での化合物の選択性を予測するのが難しくなります。構造類似化合物の場合は、保持メカニズムが類似しているため、このことはあまり問題になりません。構造的に異なる(例えばマトリックス中で)化合物の混合物を含むサンプルの場合は、より大きな課題となります。しかしながら、よりなだらかなグラジエントでは保持時間が長くなり、分離能が向上します。
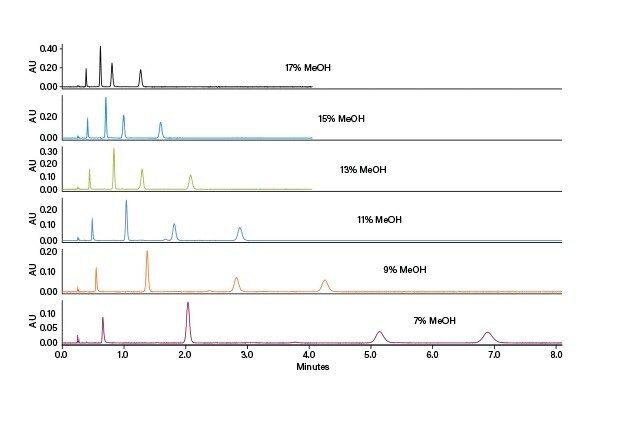
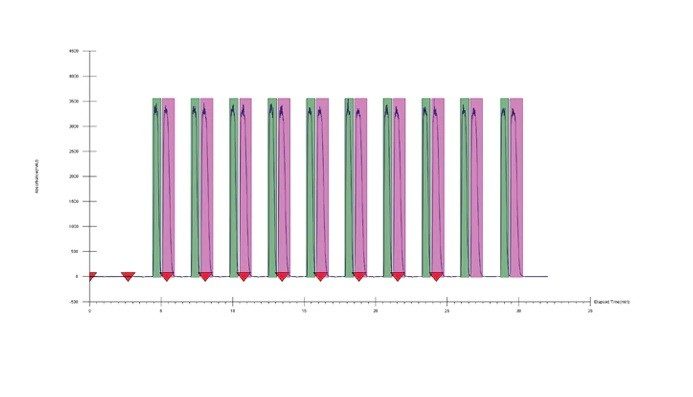
アイソクラティック条件の決定:アイソクラティック法は、スクリーニング結果に基づいて簡単に開発でき、スタックインジェクションが使用できることから、生産性が高まる理想的な方法です。保持時間、スクリーニンググラジエントの傾き、およびシステムとカラムの遅延容量の補正を使用して、溶出時の共溶媒の割合を決定することができます。SFC では通常、最適化に最適な出発点は算出された割合から 5% を引いた値です。
スクリーニンググラジエントは 5 分間で 2 ~ 20% で、グラジエント遅延は 0.46 分でした(事前に決定)。したがって、3.6%/分 と算出された傾きおよび 2% という出発割合を用いて、4.12 分における最初のピークの溶出時の共溶媒の割合を以下の式を用いて算出します:
■ 溶出時の共溶媒の割合(%) = (保持時間 – グラジエント遅延)× グラジエントの傾き + 出発点の割合(%)
■ 溶出時の共溶媒の割合(%) = (4.12 分 – 0.46 分) × 3.6%/分 + 2%
■ 溶出時の共溶媒の割合(%) = 15%
したがって、5% を引いた 10% というアイソクラティック共溶媒条件を最適化の出発点として使用しました。得られたクロマトグラフィーでは良好な分離が確認されましたが、移動相の共溶媒の割合を増やして 15% に戻しても、ピークは依然として短い実行時間で良好に分離されました。この場合、10% では負荷量の増加が、15% ではサイクル時間の短縮が可能です。