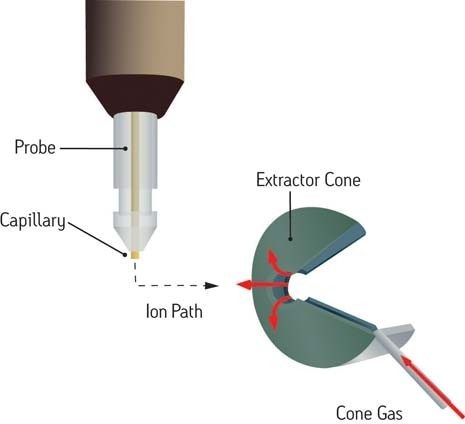
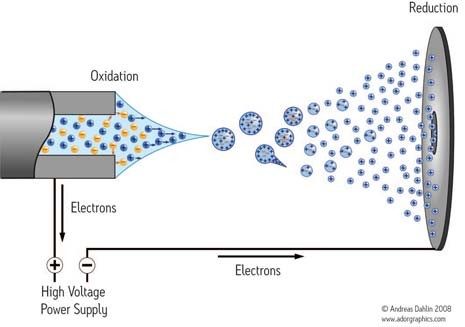
Chemische Ionisation (CI)
Moleküle, die übermäßig fragmentieren, erfordern „sanfte“ Techniken. Die chemische Ionisation (CI) erzeugt Ionen durch einen sanfteren Protonentransferprozess, der das Erscheinungsbild des Molekülions selbst bewahrt und fördert. Die Probe wird einem Überschuss an Reagenzgas ausgesetzt, wie es entsteht, wenn Methan das protonierte Molekülion (M+H) bildet. Der umgekehrte Prozess kann negative Ionen erzeugen. Durch die Übertragung des Protons auf das Gasmolekül kann in einigen Fällen das negative Ion entstehen (M-H).
Die chemische Ionisation (CI) wird manchmal für Verbindungen verwendet, deren chemische Zusammensetzung derjenigen ähnelt, die mit EI analysiert werden, um die Häufigkeit oder das Aussehen des Molekülions zugunsten einer signifikanten Fragmentierung zu erhöhen. Ähnlich wie bei der EI müssen die Proben thermisch stabil sein, da die Erwärmung in der Quelle zur Verdampfung führt. Der Ionisierungsmechanismus von CI beruht auf EI für den ersten Ionisierungsschritt, aber in der Quelle befindet sich ein chemisches Reagenzgas, wie Methan, Isobutan oder Ammoniak, unter hohem Druck. Das Reagenzgas, das in einer viel höheren Konzentration als der Analyt (R) vorhanden ist, wird durch Elektronenionisierung ionisiert, wobei primäres R+ Reagenzionen entstehen. Die Kollision von R+Ionen mit neutralen R-Molekülen führt zur Bildung stabiler Sekundärionen, die die reaktive Spezies sind, welche dann die Analytmoleküle (A) durch Ionen-Molekül-Reaktionen ionisieren.
So entsteht beispielsweise bei der Ionen-Molekül-Reaktion zwischen einem Methanion und einem Methanmolekül die recht stabile Spezies CH5+.
CH4+. + CH4 --> CH5+ + CH3.
Das reaktive Ion CH5+ kann neutrale Analytmoleküle (A) durch Protonentransfer, Hydridabstraktion oder Ladungsaustausch ionisieren.
RH+ + A --> R + AH+ (Protonenübertragung)
(R-H)+ + A --> R + (A-H)+ (Hydridabstraktion)
R+. + A --> R + A+. (Ladungsaustausch)
Die gängigsten Ionisierungsreaktionen sind die Protonierung, die für Moleküle mit Protonenaffinitäten, die höher als die des Reagenz sind, bevorzugt wird. Bei Molekülen mit geringer Protonenaffinität kommt es häufig zur Abstraktion von Hydriden und bei Reagenzien mit hoher Ionisierungsenergie zum Ladungsaustausch.
Die zu analysierende Substanz steht unter einem viel niedrigeren Druck als das Reagenzgas. Betrachtet man Methan als Reagenzgas, so bewirkt der Elektronenstoß hauptsächlich die Ionisierung des Methans. Dieses fragmentiert zum Teil auf CH3+ Diese Spezies gehen dann unter den hohen Quelldrücken Ionenmolekülreaktionen ein.
CH4+. + CH4 --> CH5+ + CH3.
CH3+ + CH4 --> C2H5+ + H2
CH5+ kann als Bronsted-Säure und C2H5+ als Lewis-Säure wirken, um Ionen aus dem Analyten zu erzeugen.
Eine sorgfältige Auswahl des CI-Reagenzgases kann die Ladungsübertragung auf ein Analytmolekül verbessern, da der Säuregrad der Gasphase des chemischen Ionisierungsgases die Effizienz der Ladungsübertragung beeinflusst. Bei der CI ist es wahrscheinlicher, dass der Analyt zu einem Molekülion führt, wobei durch die geringere Fragmentierung die Energie erhalten bleibt, die normalerweise bei der EI zum Aufbrechen von Bindungen internalisiert wird.