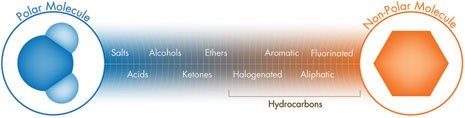
Wenn schwache Ionenaustauscher neutralisiert werden, können sie Spezies durch hydrophobe [Umkehrphase] oder hydrophile [Normalphasen] Wechselwirkungen zurückhalten und trennen; in diesen Fällen wird die Elutionsstärke durch die Polarität der mobilen Phase bestimmt [Abbildung R-1]. Daher können schwache Ionenaustauscher für Mixed-Mode-Trennungen verwendet werden [Trennungen basieren sowohl auf Polarität als auch auf Ladung].
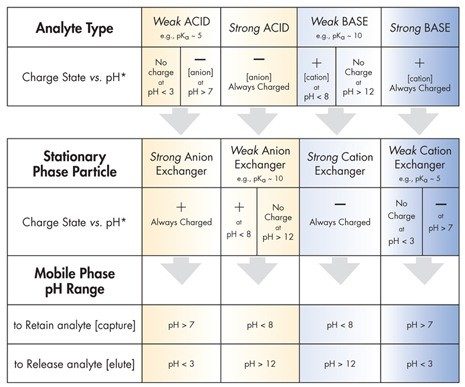
Tabelle D enthält Richtlinien für die Hauptkategorien des Ionenaustauschs. Um zum Beispiel einen stark basischen Analyten [immer positiv geladen] zurückzuhalten, verwenden Sie einen schwachen Kationenaustauscher-Partikel der stationären Phase bei einem pH-Wert > 7; dies gewährleistet eine negativ geladene Partikeloberfläche. Um die starke Base freizusetzen oder zu eluieren, senken Sie den pH-Wert der mobilen Phase auf unter 3; dadurch wird die Oberflächenladung entfernt und der Ionenaustausch-Retentionsmechanismus abgeschaltet.
Beachten Sie, dass ein pKa der pH-Wert ist, bei dem 50 % der funktionellen Gruppe ionisiert und 50 % neutral sind. Um einen im Wesentlichen neutralen oder eine vollständig geladene Analyt- oder Partikeloberfläche zu gewährleisten, muss der pH-Wert auf einen Wert eingestellt werden, der mindestens 2 Einheiten über dem pKa-Wert liegt [in Tabelle D angegeben].
Verwenden Sie keinen starken Kationenaustauscher, um eine starke Base aufrechtzuerhalten; beide bleiben geladen und ziehen sich stark an, was eine Elution der Base nahezu unmöglich macht. Sie kann nur entfernt werden, indem der starke Kationenaustauscher mit einer konkurrierenden Base überlagert wird, die eine noch stärkere Retention zeigt und die interessierende Verbindung verdrängt, indem sie die Konkurrenz um die aktiven Austauschstellen gewinnt. Dieser Ansatz ist bei HPLC und SPE selten praktisch oder sicher. [Die Arbeit mit sehr starken Säuren und Basen ist gefährlich und sie können die Konstruktionsmaterialien von HPLC-Flüssigkeitssystemen angreifen!]