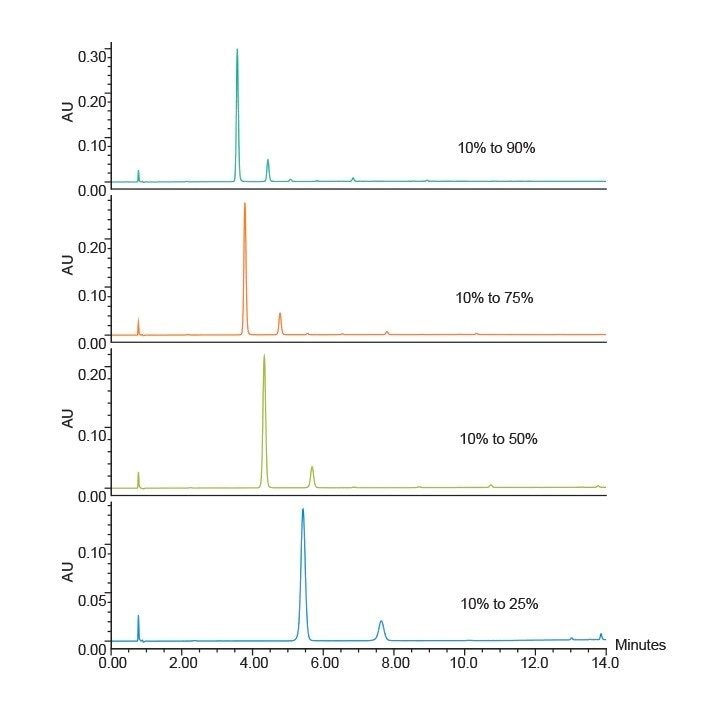
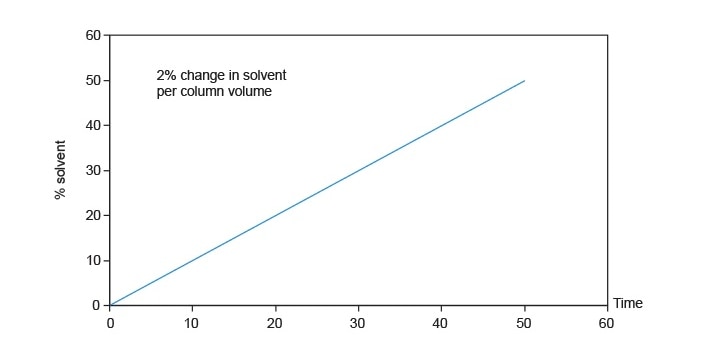
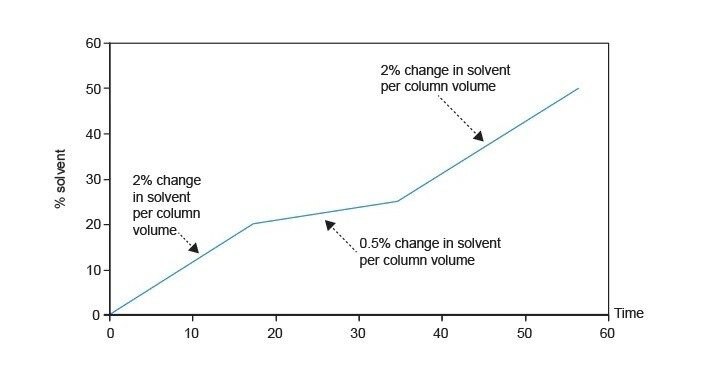
Développement d’un gradient ciblé
Les calculs suivants peuvent être utilisés pour générer un gradient ciblé pour le composé d'intérêt après l'analyse de l'échantillon avec un gradient de référence rapide. Chaque équation est suivie d'un exemple théorique.
Il faut tout d'abord déterminer le volume mort du système pour l'instrument utilisé lors de l'analyse du gradient de référence. Des instructions pour la détermination du volume mort sont disponibles dans la section du présent guide technique intitulée « Détermination du volume mort » ou dans l'outil applicatif « Analytical to Prep Gradient Calculator » (Calculateur de gradient analytique à préparatif) sur www.waters.com/prepcalculator.
En outre, il faut également déterminer le volume de la colonne utilisée pour l'analyse de référence. Il peut être calculé en utilisant le volume d'un cylindre V = πr²h et en compensant le volume occupé par le matériau de conditionnement c'est-à-dire V = r²h (66 %). L'outil applicatif « Analytical to Prep Gradient Calculator » (Calculateur de gradient analytique à préparatif) peut également être utilisé pour effectuer ce calcul.
Équation 1 : volume de la colonne
Volume de la colonne = π r² x hauteur x 66 % disponible pour la phase mobile/1 000 pour convertir les mm³ en mL
Exemple :
VC = 3,14 x (4,6 mm/2)² x 50 mm x 0,66/1000
VC = 548 mm³ = 0,548 mL
Équation 2 : volume de décalage entre la formation du gradient et le détecteur
Volume de décalage = volume du système* + volume de la colonne
*Volume du système ou volume mort
Exemple :
Volume de décalage = 1,04 mL + 0,548 mL
Volume de décalage = 1,588 mL
Équation 3 : temps jusqu’au détecteur
Temps jusqu’au détecteur = décalage en mL/débit en mL/min
Exemple :
Temps jusqu’au détecteur = 1,588 mL/1,5 mL/min
Temps jusqu’au détecteur = 1,06 min
Équation 4 : temps de formation de la concentration d’élution
Temps de la concentration d’élution = Temps de rétention du pic d’intérêt – Temps jusqu’au détecteur – Maintien du gradient
Exemple :
Temps de la concentration d’élution = 6,0 min – 1,06 min – 0,0 min
Temps de la concentration d’élution = 4,94 min
Équation 5 : concentration d’élution en pourcentage
Concentration d’élution en pourcentage = temps de la concentration d’élution/durée du segment de gradient de référence x changement de référence + pourcentage du gradient de référence initial
Exemple :
Concentration d’élution en pourcentage = 4,94 min/6,0 min x 90 % + 5 %
Concentration d’élution en pourcentage = 79,1 %
Équation 6 : nombre de volumes de colonne (VC)
Nombre de volumes de colonne = 1 volume de colonne/mL × débit mL/min × durée du segment de gradient de référence
Exemple :
Nombre de VC = 1 VC/0,548 mL x 1,5 mL/min x 6 min
Nombre de VC = 16,4
Équation 7 : pente du gradient de référence
Pente du gradient de reconnaissance = pourcentage de variation du gradient de référence/nombre de volumes de colonne
Exemple :
Pente du gradient de référence = (95 % – 5 %)/16,4 VC
Pente du gradient de référence = 5,49 %/VC
Équation 8 : pente du gradient ciblé
Pente du gradient ciblé = 1/5 x pente du gradient de référence
Exemple :
Pente du gradient ciblé = 1/5 x 5,49 %/VC
Pente du gradient ciblé = 1,1 %/VC
Le segment de gradient ciblé est créé entre 5 % en dessous et 3 à 5 % au-dessus du pourcentage d'élution estimé. À titre d'exemple, le pourcentage d'élution est de 79 %.
Équation 9 : durée du segment de gradient ciblé
Durée du segment de gradient ciblé = plage en % x (1/pente du gradient de référence) x volume de colonne x (1/débit)
Exemple :
Durée du segment de gradient ciblé = (79 % + 5 %) - (79 % - 5 %) x (1/1,1 %) x (0,548 mL) x (1/1,5 mL/min)
Durée du segment de gradient ciblé = 3,32 min
La dernière étape consiste à écrire le gradient ciblé en utilisant les valeurs calculées dans les formules précédentes.