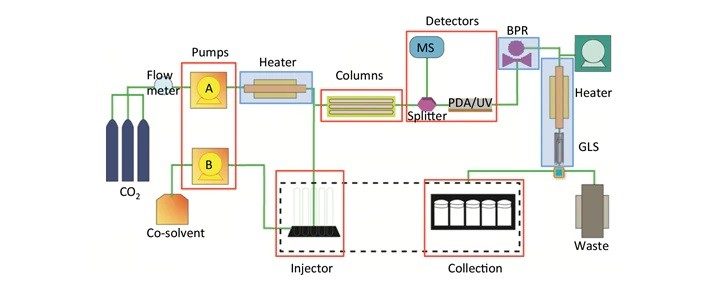
有关各组件的注意事项
泵送:SFC和LC一样,都采用二元泵系统输送溶剂“A”(对于SFC而言始终为CO2)和溶剂“B”(通常为极性有机溶剂,如甲醇)。CO2可按体积泵送(体积流量,分析级分离常用),也可按质量泵送(质量流量,制备级分离常用),具体取决于分离的规模。由于CO2可压缩,因此等体积CO2未必等质量(密度差异)。只要流速可不受环境条件影响保持重现性,上述两种输送方式均有价值。大多数情况下,CO2会在泵之前或者泵头处冷却,然后以液体形式泵送,目的是减少流动相密度偏差。另外,所有密封件、单向阀、管路和接头都必须能承受可压缩高压流体,不能泄漏。由于CO2极易通过任何开口扩散,即使少量泄漏也会显著影响系统性能和色谱分离。无论是管路泄漏、密封件还是单向阀造成的CO2泵送问题,不仅会改变保留时间和选择性,还会影响压力(密度)控制和基线噪音。
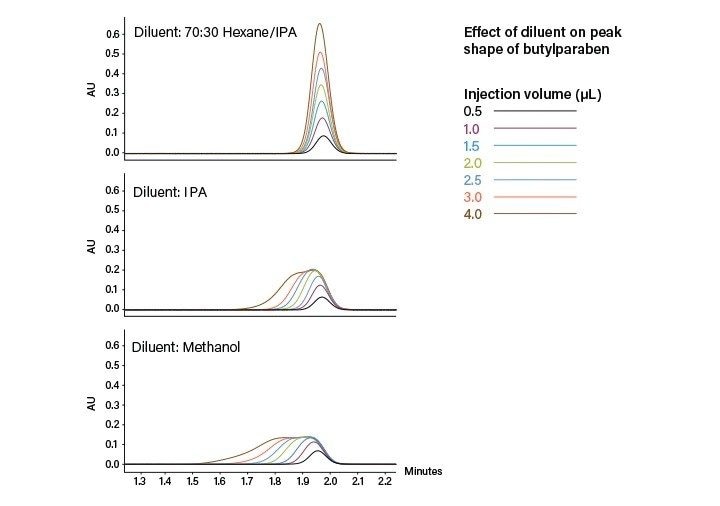
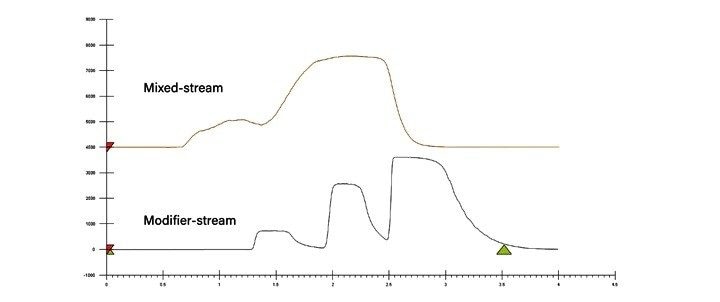
进样:SFC有混合流和改性剂流两种常用进样模式,我们将在“SFC的进样策略”部分详细介绍。混合流SFC进样方案包括降压步骤,其目的是在上样之前排空定量环中的所有CO2。设有混合流和改性剂流两种进样模式的系统有时也会将降压步骤用作安全预防措施。降压操作通常由二级排放阀完成。这种进样设置有一个缺点,即进样时增加了系统的柱外体积,这可能会导致峰展宽和分离度损失。使用小内径管路尽可能减小柱外体积,有助于减小峰展宽。
柱温箱和加热:在SFC中,压力和温度是影响分离性能的方法参数,因为它们会影响流动相密度。因此,我们必须对流动相和色谱柱进行适当的加热和温度控制。如果温度控制不当,色谱柱温度梯度会对峰形和分离度产生不利影响。大多数SFC系统通过在柱温箱中加热色谱柱、预加热流动相或将这两种方法联用来控制温度。
正确选用色谱柱填料(手性和非手性)对SFC至关重要,因此色谱柱筛选是SFC应用的必要步骤。许多SFC系统的柱温箱内都配有切换阀,供用户在多根色谱柱之间切换。当需要不同色谱柱填料来纯化多种不同的样品和目标物质时,色谱柱筛选尤为重要。
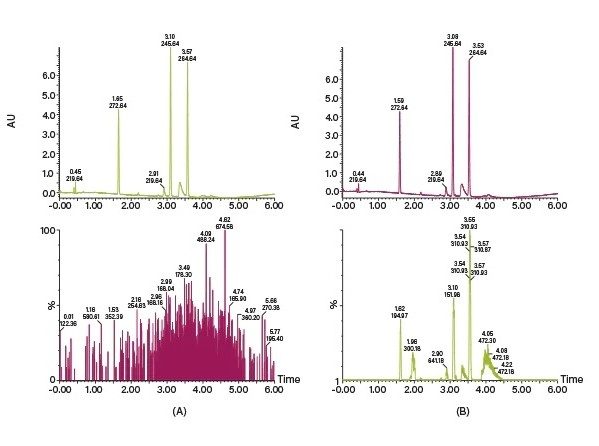
检测器:SFC可兼容纯化会用到的所有典型检测技术,例如UV/Vis、PDA、MS和ELS,而且可在同一系统中使用多个检测器。一般将UV/Vis和PDA检测器连入主流路,因为它们是非破坏性检测器,被用于初级检测。应根据SFC所采用的压力来确定上述检测器的流通池规格。MS和ELS等破坏性检测器配合分流器接入,分流器可对分流比进行相对控制,并通过调整溶剂来优化检测信号。系统可记录或使用多个检测器的信号(或通道)来触发馏分收集,有效扩展了分析型和制备型SFC技术的应用范围。上述检测器在SFC中的应用将在本章“SFC中的光学和MS检测”部分进行详细介绍。
背压调节器(BPR):控制流经色谱柱的流动相密度是SFC仪器设计的关键之一,因为所有化合物的溶解性和保留因子都与流动相的密度密切相关。密度控制主要通过控制系统压力实现。背压调节器是专用于控制系统柱后压力(背压),使其维持方法指定压力的自动化设备。要实现稳定、可重现的色谱分离,柱后压力必须保持恒定(保持设定值),即便在梯度条件下和不同运行之间也应如此。
尽管我们认为CO2在高于74 bar(约1073 psi)和31 °C时就会转变为超临界状态,但一般不建议在接近临界点的条件下运行系统,因为此时温度或压力发生极小的变化都会显著改变CO2密度。因此,就保留时间和分离度而言,在该范围内开发的方法稳定性较差。另外,如果使用助溶剂,则低压条件下更容易发生相分离,进而导致基线噪音。所以大部分SFC方法都使用较高的压力设定值,一般在100 bar (1450 psi)以上。
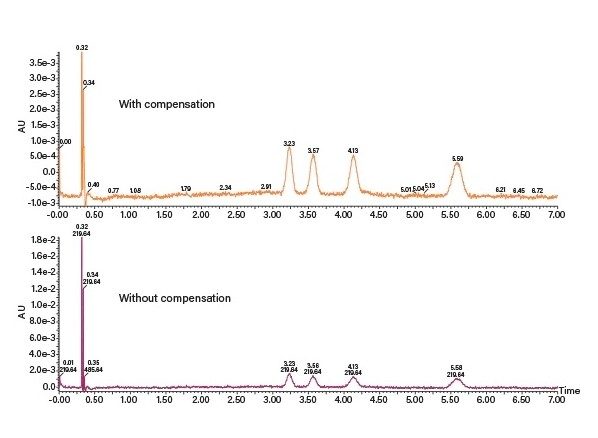
收集触发器:Prep SFC的馏分收集可通过阈值、时间和斜率收集模式在多个通道和检测器处进行(具体定义见表3)。系统运用布尔逻辑执行更加智能的收集,这种收集要求同时满足多个条件才能触发。收集条件可以是模式的组合,例如阈值和时间,也可以是检测器信号的组合,例如UV阈值和质量数确证数据。