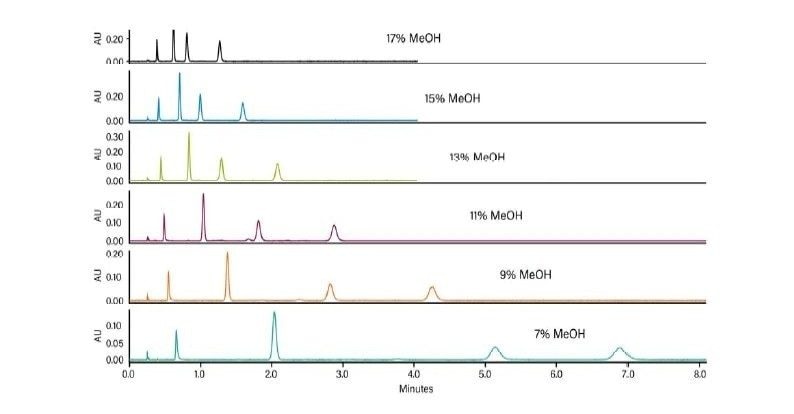
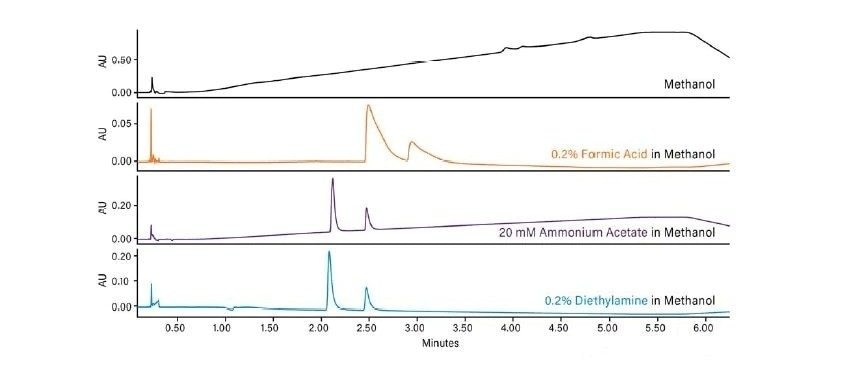
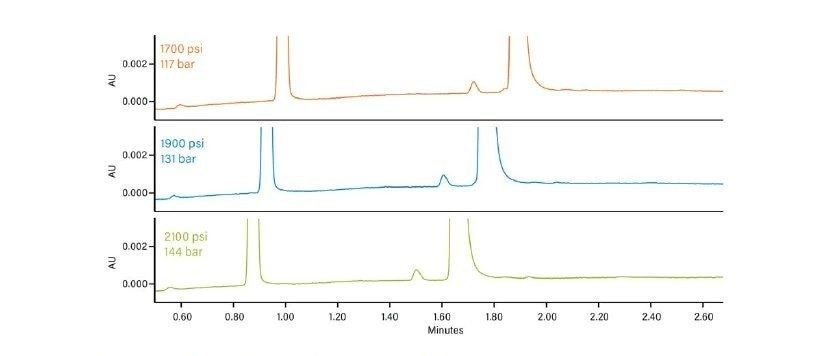
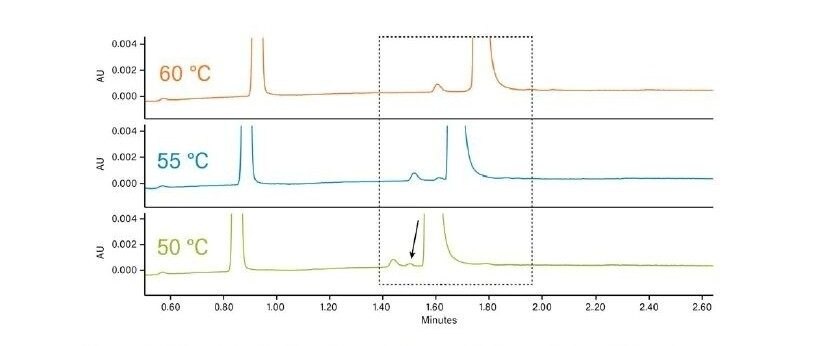
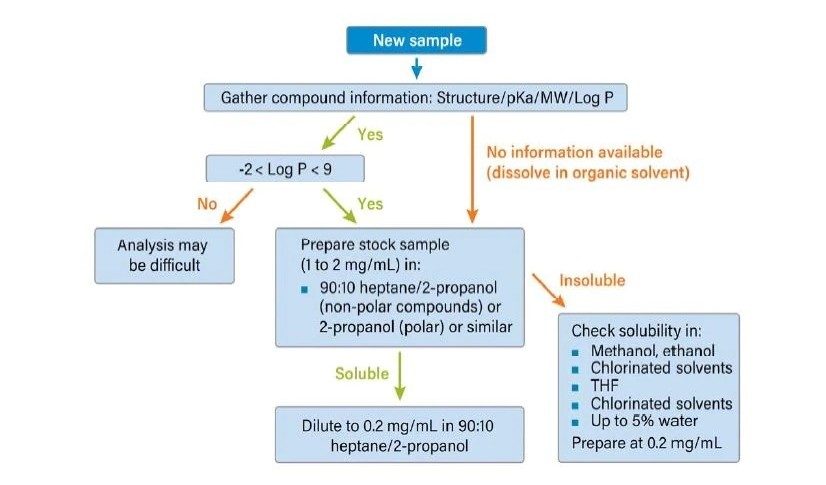
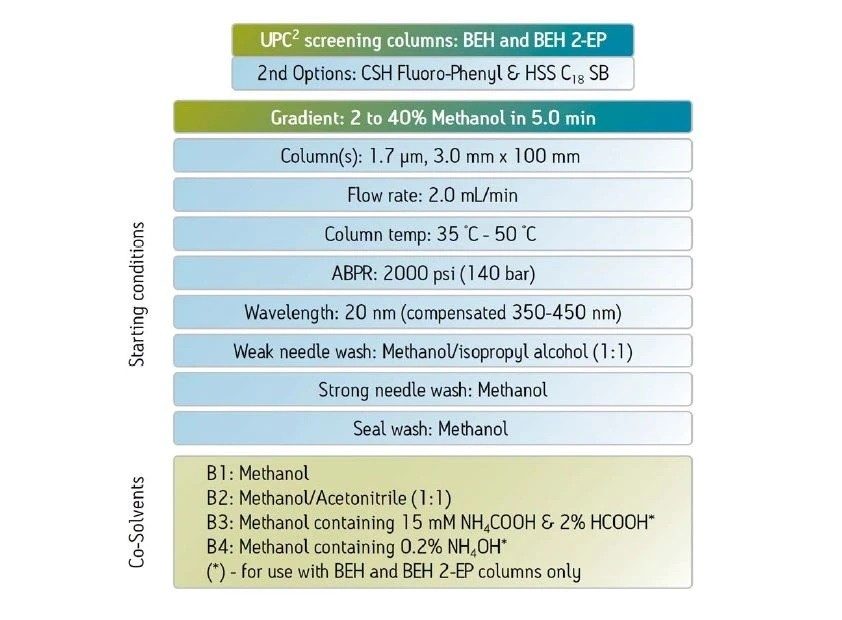
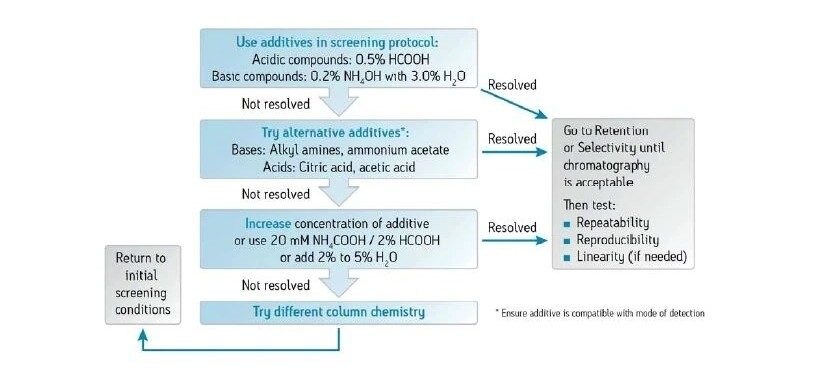
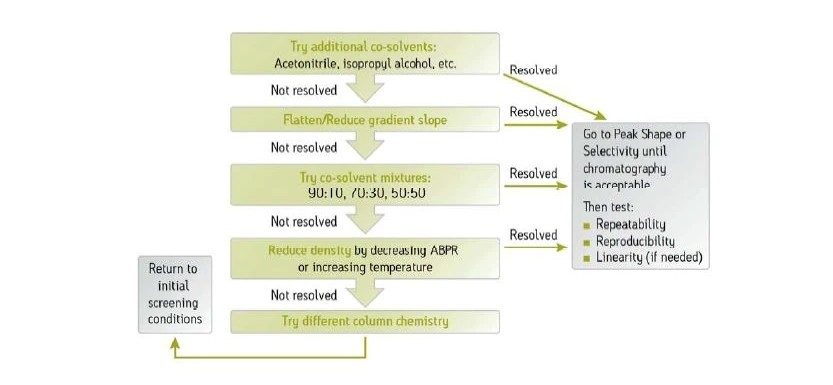
Abbildung 36 zeigt ein Protokoll zur Erhöhung der Retention. Wie bei jeder Form der Flüssigchromatographie besteht die erste Möglichkeit darin, ein anderes (schwächeres) Hilfslösungsmittel zu verwenden. Methanol ist das stärkste Hilfslösungsmittel, das bei der CC verwendet wird; die Verwendung eines schwächeren Hilfslösungsmittels, wie z. B. Acetonitril oder eines anderen Alkohols, verlängert die Retentionszeit auf der Säule. Das Abflachen oder Verringern der Steigung des Gradienten (Verringern des endgültigen Prozentsatzes des Hilfslösungsmittels oder Erhöhen der Gradientendauer) kann ebenfalls wirksam sein. Das Mischen von Hilfslösungsmitteln und die Verringerung der Methanolkonzentration schwächt die Gesamtstärke des Hilfslösungsmittels, wodurch die Retention erhöht wird. Die Möglichkeit, die Dichte der mobilen Phase in der Säule zu manipulieren, ist eine einzigartige Eigenschaft der SFC und der CC. Dadurch ändert sich die Gesamtretention, da eine niedrigere Dichte einer höheren Retention entspricht. Dies erfolgt durch Verringern der ABPR-Einstellung und/oder Erhöhen der Temperatur. Schließlich ist die Verwendung einer anderen Säulenchemie eine weitere Strategie zur Erhöhung der Retention.
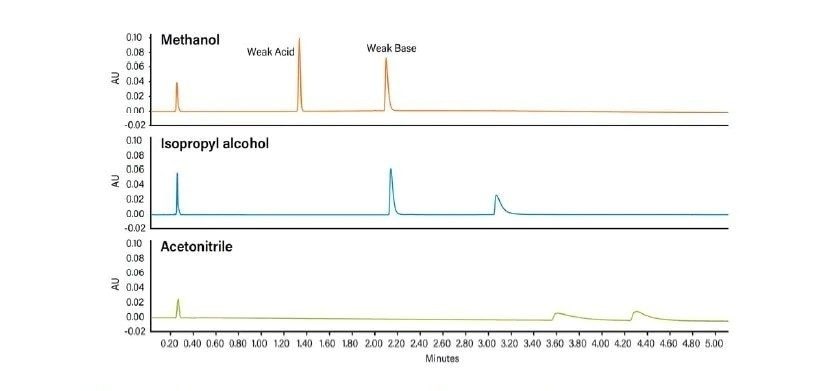
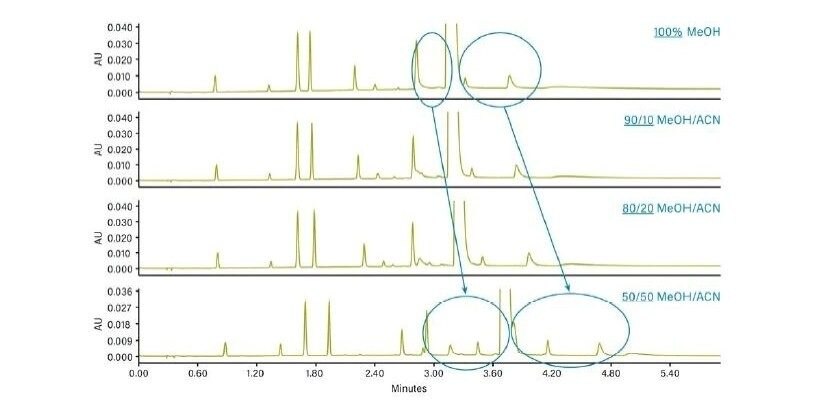
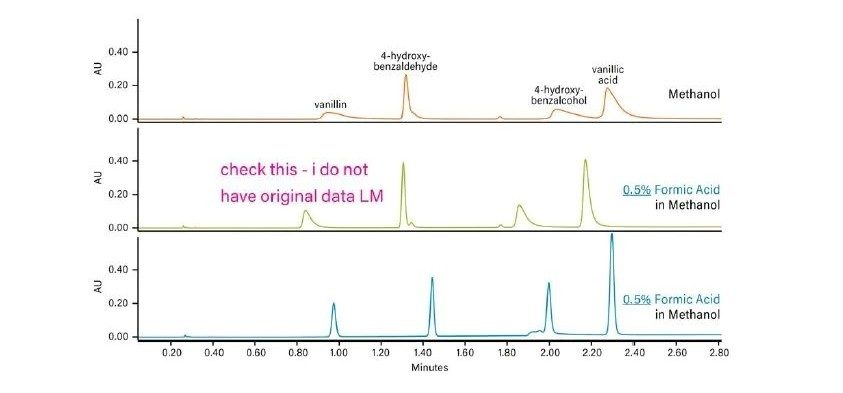
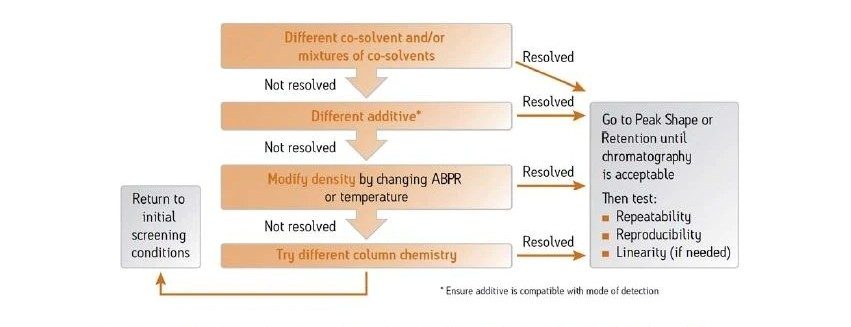
Abbildung 37 zeigt ein Protokoll zur Änderung der Selektivität (Elutionsreihenfolge, relative Retention) der Trennung. Ein Ansatz ist der Versuch, andere Hilfslösungsmittel zu verwenden, z. B. Acetonitril anstelle von Methanol. Der Wechsel von einem alkoholischen, protischen Hilfslösungsmittel zu einem nicht-alkoholischen, aprotischen Hilfslösungsmittel wie Acetonitril hat einen viel größeren Einfluss auf die Selektivität als der Wechsel von Methanol zu einem anderen Alkohol. Da eine Verringerung der Methanolkonzentration die Gesamtstärke des Hilfslösungsmittels schwächt, kann dies die Retention erhöhen und die Selektivität verändern. Die Manipulation der Dichte der mobilen Phase kann auch die gesamte Analytretention verändern, da diese Dichteeffekte für spezifische Analyten variieren können, was ausreichen kann, um eine gegebene Trennung zu optimieren. Schließlich empfiehlt dieses Protokoll gegebenenfalls eine andere Säulenchemie.