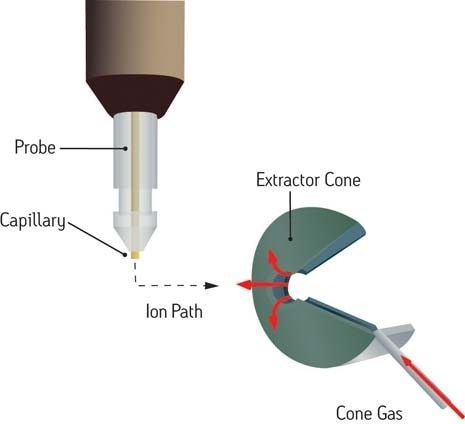
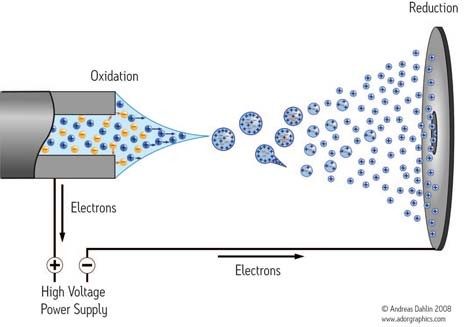
Ionisation chimique
Les molécules qui se fragmentent excessivement nécessitent d’employer des techniques douces. L’ionisation chimique, ou CI, produit des ions par un processus de transfert de protons plus doux, qui favorise l’apparition de l’ion moléculaire et le préserve. L’échantillon est exposé à un excès de gaz réactif, tel que celui qui se dégage lorsque le méthane forme l’ion moléculaire protoné (M+H). Le processus inverse permet de produire des ions négatifs. Le transfert du proton à la molécule de gaz peut, dans certains cas, produire un ion négatif (M-H).
L’ionisation chimique (CI) est parfois utilisée pour les composés ayant une chimie proche de ceux analysés par EI afin d’améliorer l’abondance ou l’apparition de l’ion moléculaire en faveur d’une fragmentation significative. Comme pour l’EI, les échantillons doivent être thermiquement stables, car ils seront vaporisés par le chauffage de la source. Le mécanisme d’ionisation de la CI est similaire à celui de l’EI pour l’étape d’ionisation initiale à la différence que la source contient un gaz réactif chimique, tel que le méthane, l’isobutane ou l’ammoniac, à haute pression. Le gaz réactif (R), présent à une concentration beaucoup plus élevée que l’analyte, est ionisé par ionisation électronique pour donner les ions réactifs R+. primaires. La collision des ions R+. avec les molécules R neutres entraînent la formation d’ions secondaires stables, qui sont les espèces réactives qui ionisent les molécules d’analyte (A) par réaction ion-molécule.
Par exemple, la réaction ion-molécule entre un ion méthane et une molécule de méthane produit l’espèce CH5+, qui est relativement stable.
CH4+. + CH4 --> CH5+ + CH3.
L’ion réactif CH5+ peut ioniser les molécules d’analyte neutres (A) par transfert de protons, abstraction d’hydrure ou échange de charge.
RH+ + A --> R + AH+ (transfert de proton)
(R-H)+ + A --> R + (A-H)+ (abstraction d’hydrure)
R+. + A --> R + A+. (échange de charge)
La réaction d’ionisation la plus répandue est la protonation. On la retrouve pour les molécules ayant une affinité protonique supérieure à celle du réactif. L’abstraction d’hydrure est courante pour les molécules à faible affinité protonique, tandis qu’un échange de charges se produit avec les réactifs à haute énergie d’ionisation.
La substance à analyser est à une pression bien inférieure à celle du gaz réactif. Si l’on utilise le méthane comme gaz réactif, l’impact électronique provoque principalement l’ionisation du méthane. Celui-ci se fragmente en partie en CH3+. Ces espèces subissent ensuite des réactions ion-molécule à pression élevée dans la source.
CH4+. + CH4 --> CH5+ + CH3.
CH3+ + CH4 --> C2H5+ + H2
Le CH5+ peut agir comme un acide de Brönsted et le C2H5+ comme un acide de Lewis pour produire des ions à partir de l’analyte.
Le choix du bon gaz de réactif pour la CI peut améliorer le transfert de charge vers une molécule d’analyte, car l’acidité de la phase gazeuse du gaz d’ionisation chimique influe sur l’efficacité du transfert de charge. En CI, l’analyte est plus susceptible de produire un ion moléculaire avec une fragmentation réduite, ce qui permet de conserver l’énergie normalement internalisée dans l’EI pour rompre les liaisons.