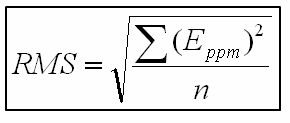Comparaison de la fidélité d’un instrument à l’autre : unités de millimasse (mmu), erreur de mesure (ppm) et résolution
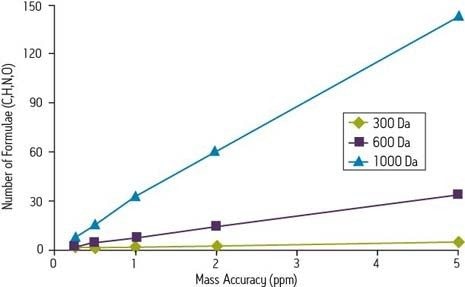
Selon le document Accurate Mass Best Practice Guide du programme VIMMS, une initiative du National Measurement System du Royaume-Uni, la plupart des instruments utilisés pour les mesures en masse exacte peuvent atteindre une fidélité minimale de 10 ppm.
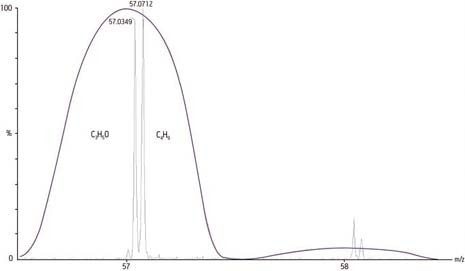
Mesurée par un spectromètre de masse actuel avec une exactitude de 2 mmu, une masse calculée de 118 Da afficherait une erreur de 17 ppm, suffisante selon les normes actuelles pour déterminer sans ambiguïté une formule chimique de cette masse :
Masse exacte monoisotopique calculée = 118 Da
Masse exacte mesurée = 118,002 Da
Différence = 0,002 mmu
Erreur [Différence/masse exacte calculée x 106] = 17 ppm
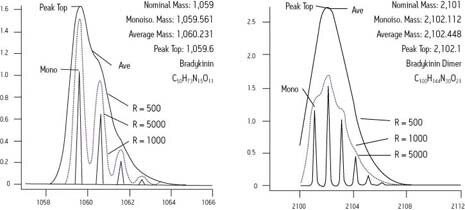
Un instrument capable de mesurer une réponse à m/z 750, également avec une erreur de 2 mmu, aurait une exactitude de 2,7 ppm. Dans le premier cas, la mesure est plus que suffisante pour l’identification sans ambiguïté d’une formule chimique, selon les normes publiées dans le Journal of The American Society for Mass Spectrometry. Mais dans le dernier cas, l’exactitude de la mesure est insuffisante. Seule la spectrométrie de masse à résonance cyclotron ionique (FTICR) la plus performante permet d’atteindre une telle fidélité à des masses plus élevées.
Une méthode complète d’évaluation de la capacité de l’appareil à mesurer des masses exactes, qui s’apparente à son usage prévu, consiste à calculer l’erreur quadratique moyenne ou RMS. À des fins d’illustration, ce qui suit est adapté des caractéristiques d’exactitude de la mesure de masse d’un spectromètre de masse Tof disponible dans le commerce.
« Dans des conditions d’utilisation normales, l’exactitude de la mesure de masse de l’instrument sera meilleure qu’une erreur RMS donnée en ppm sur la plage m/z donnée, sur la base d’un nombre de mesures répétées consécutives d’un pic d’analyte (de m/z donné), en utilisant un pic de référence adapté (de m/z donné). Les pics de l’analyte et de référence doivent avoir une intensité suffisante et être exempts d’interférences provenant d’autres masses. »
Certains points et hypothèses importants doivent être pris en compte :
- L’instrument a déjà été étalonné avec des pics de masse connue à l’aide d’une solution étalon. Le pic de référence permet de prendre en compte toute variation de l’étalonnage de l’instrument dans le temps, et l’exactitude de la mesure de masse est déterminée à l’aide du pic d’analyte.
- Les conditions normales d’utilisation peuvent également inclure des détails sur les conditions chromatographiques (pour les caractéristiques de performance de la LC/MS) et toutes les conditions d’utilisation associées au spectromètre (par exemple, résolution en masse, m/z d’intérêt ou fréquence d’acquisition spectrale).
- Intensité suffisante : suppose que le nombre d’ions ne nuit pas à la caractérisation de l’exactitude (de la mesure de la masse) et de la fidélité de l’instrument en question. Un défaut d’ions nuit aux statistiques ioniques, tandis qu’un excès peut entraîner une saturation du détecteur. Ces deux phénomènes entraînent une variation plus importante de l’écart-type des mesures répétées et nuisent au calcul de l’erreur quadratique (vaut également pour l’étalonnage de l’instrument).
- Exempt d’interférences : suppose que la mesure de la masse du pic de masse connue est exempte d’interférences dues à des ions de masse identique ou similaire. Le chevauchement des pics diminue l’exactitude de la mesure de masse, ce qui nuit également à la caractérisation correcte de l’exactitude ou de la fidélité de l’instrument (vaut également pour l’étalonnage de l’instrument).
- La référence est une bonne représentation de la plage m/z pertinente pour l’analyse d’un type d’échantillon donné.
L’erreur RMS est calculée à l’aide de la formule suivante, où Eppm est l’erreur en ppm et n le nombre de masses prises en compte :