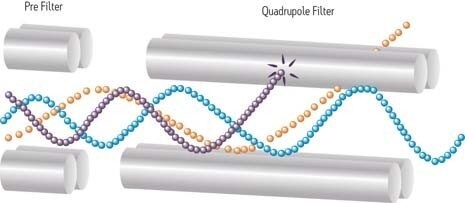
Un piège à ions fonctionne selon des principes similaires à ceux d’un instrument quadripolaire. En revanche, contrairement au quadripôle, qui filtre les ions en circulation, le piège à ions et le cyclotron ionique (ICR), plus performant que le premier, stockent les ions dans un espace tridimensionnel. Avant la saturation, le piège ou le cyclotron peut expulser certains ions vers le détecteur, en fonction de leur masse. Il est possible de réaliser une série d’expériences dans l’enceinte du piège, en fragmentant un ion d’intérêt de sorte à mieux définir le précurseur par ses fragments. Les champs générés par les tensions RF appliquées à une géométrie empilée ou en « sandwich » (électrodes-chapeaux à chaque extrémité) piègent les ions dans l’espace situé entre les deux électrodes. L’augmentation ou le balayage de la tension RF expulse les ions de leur fréquence séculaire ou du piège. La gamme dynamique est parfois limitée. Les limites de volume et de capacité à accueillir des ions restreignent la portée de l’instrument, en particulier pour les échantillons en matrice complexe.
Les pièges à ions ont été introduits dans les années 80. Mais les limites imposées par le schéma d’ionisation interne utilisé dans ces premiers instruments les rendaient inadaptés pour de nombreuses applications. Ce n’est qu’avec l’avènement de l’ionisation externe que les instruments sont devenus plus polyvalents.
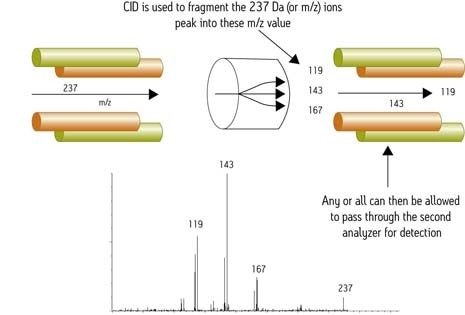
La possibilité d’effectuer une fragmentation séquentielle pour dériver plus d’informations structurales à partir d’un analyte unique (c’est-à-dire fragmenter un ion, sélectionner un fragment particulier et répéter le processus) est appelée MSn. Les pics chromatographiques en GC ne sont pas suffisamment larges pour permettre plus d’une fragmentation (MS/MS ou MS2). Contrairement aux instruments quadripolaires et à secteur, les instruments à piège ionique réalisent les expériences de MS/MS ou de fragmentation dans le temps plutôt que dans l’espace. Ils ne sont donc pas adaptés à certaines expériences MS/MS comme les comparaisons de perte de neutre et d’ions précurseurs. De plus, lors d’utilisation d’un piège à ions en mode MS/MS, le tiers inférieur du spectre MS/MS est perdu, en raison de la conception du piège. Pour contrer cette perte, certains fabricants proposent, via leur logiciel, des exigences de balayage plus étendues qui nécessitent de changer les paramètres de fonctionnement au cours de l’acquisition des données.
La conception du piège plafonne le ratio entre le rapport masse/charge (m/z) d’un ion précurseur et celui de l’ion fragment piégé le plus faible. Ce phénomène est communément appelé « règle du tiers ». Par exemple, les ions fragments issus d’un ion à m/z 1 500 ne seront pas détectés en dessous de m/z 500, ce qui constitue une limite significative pour le séquençage peptidique de novo. Le piège à ions a une gamme dynamique limitée, du fait des effets de charge d’espace lorsqu’un trop grand nombre d’ions pénètrent dans l’espace de piégeage. Les fabricants ont mis au point un balayage automatisé, qui compte les ions avant qu’ils n’entrent dans le piège, limitant ainsi le nombre autorisé d’ions. Des difficultés peuvent encore survenir lorsque la quantité d’un ion d’intérêt est relativement faible par rapport aux ions de la matrice.
En raison de leur similarité, certains instruments quadripolaires ont été conçus pour intégrer les avantages du quadripôle linéaire et du piégeage des ions. Ces instruments hybrides offrent une meilleure sensibilité et permettent de réaliser des expériences à la volée inaccessibles avec l’une ou l’autre des techniques employée seule. De tels instruments sont parfois appelés pièges linéaires ou Q-traps. L’augmentation du volume d’un piège linéaire (par rapport à un piège à ions tridimensionnel) offre une gamme dynamique plus étendue.
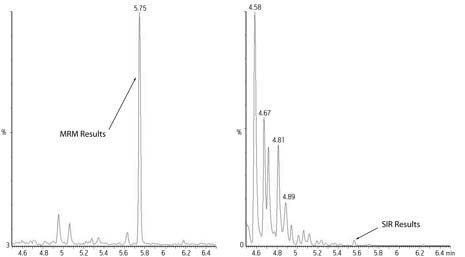
Contrairement aux instruments quadripolaires, les instruments à piégeage ionique n’effectuent pas de balayage. Les modes SIM (suivi d’un ion unique) ou SIR (fragmentogramme) n’améliorent donc pas la sensibilité des pièges à ions comme sur les instruments à quadripôle et à secteur.
Les instruments à résonance cyclotron ionique à transformée de Fourier (FTICR) offrent des capacités exceptionnelles de mesure de la masse et permettent de résoudre des masses très proches. Bien qu’ils s’avèrent peu pratiques pour la plupart des applications, équipés d’un aimant de 14,5 tesla, ils peuvent atteindre une résolution supérieure à 3,5 millions et ainsi distinguer deux entités moléculaires ayant une différence de masse inférieure à la masse d’un électron.
Les instruments à cyclotron piègent les ions par voie électrostatique dans une cellule par le biais d’un champ magnétique constant. Les impulsions de tension RF créent un mouvement ionique orbital. Les ions en orbite génèrent alors un petit signal au niveau des plaques de détection de la cellule (la fréquence orbitale de l’ion). La fréquence est inversement proportionnelle au rapport m/z des ions, et l’intensité du signal est proportionnelle au nombre d’ions de même valeur m/z dans la cellule. À des pressions de cellule très basses, un instrument à cyclotron peut maintenir l’orbite d’un ion pendant de longues périodes, offrant ainsi des mesures hautement résolutives.
La dissociation induite par collision à irradiation soutenue sans résonance (SORI-CID) est une technique de CID utilisée en spectrométrie de masse à résonance cyclotron ionique à transformée de Fourier. Les ions sont accélérés par un mouvement cyclotronique où une pression croissante provoque des collisions produisant des fragments. Après la fragmentation, la pression est réduite et le vide est rétabli pour analyser les ions fragments.
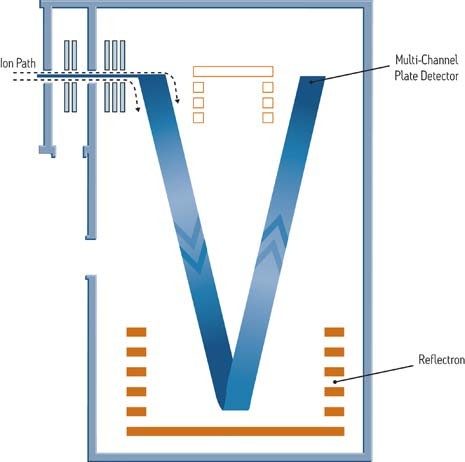
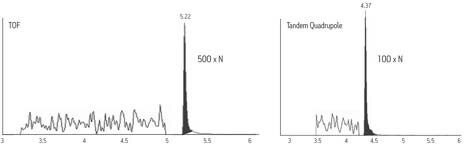
Bien que développés il y a de nombreuses années, les instruments à temps de vol (ToF) sont aujourd’hui à la base de nombreux travaux en raison de leur système électronique rapide et précis et des techniques d’ionisation modernes qu’ils emploient comme l’ESI. Un instrument à temps de vol fournit une mesure de la masse exacte à quelques parties par million (ppm) près de la masse réelle d’une molécule. Analyseur de masse à dispersion temporelle, l’instrument à temps de vol est utilisé de façon linéaire ou comme un réflectron à l’aide de grilles électrostatiques et de lentilles. Lorsqu’il est utilisé comme un réflectron, la résolution est augmentée sans perte significative de sensibilité ni augmentation de la taille du tube de vol (ou de dérive).