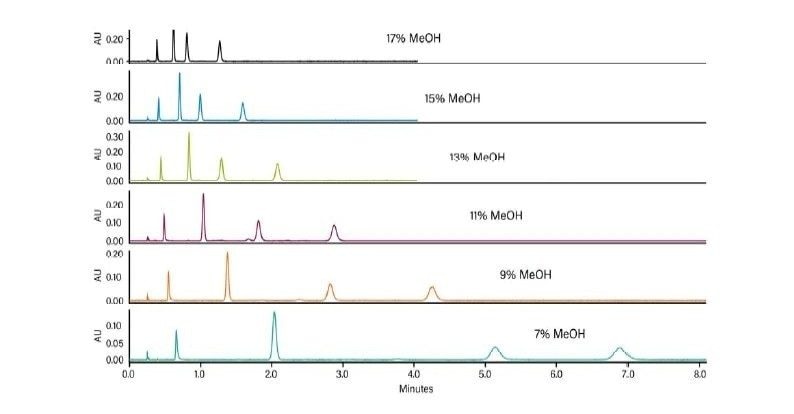
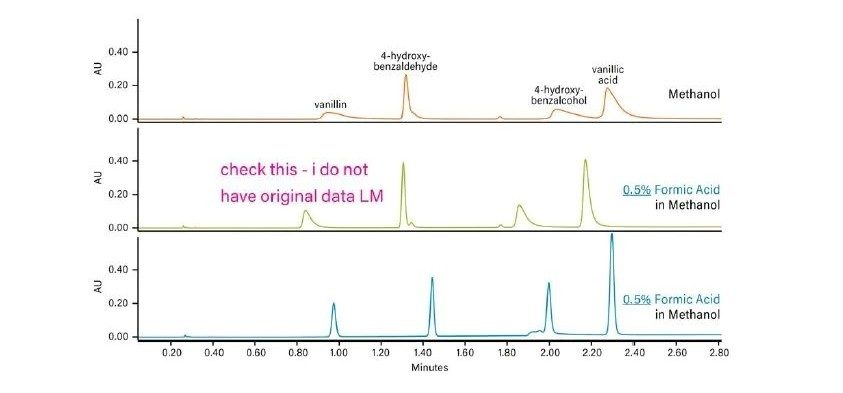
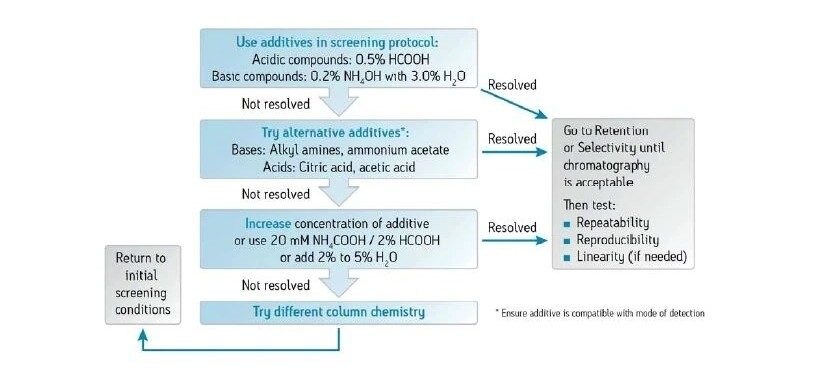
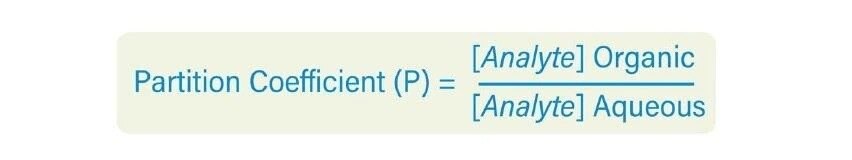Les cosolvants ajoutés à la phase mobile de CO2 diminuent généralement le temps de rétention d'un analyte. Lorsque la concentration en cosolvant augmente, la polarité de la phase mobile change, ce qui diminue le ou les temps de rétention. La Figure 25 illustre l'effet d'un changement de concentration en cosolvant sur la rétention dans une séparation isocratique. Lorsque la concentration du cosolvant d'élution fort (méthanol) diminue, la rétention des analytes augmente. Il s’agit d’un phénomène identique à celui observé en RPLC.
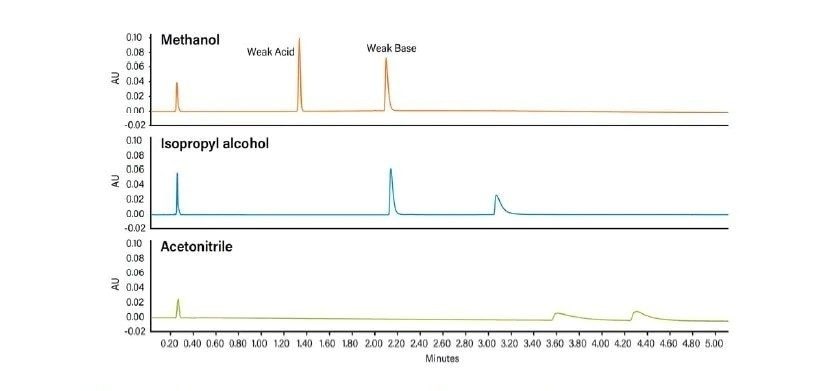
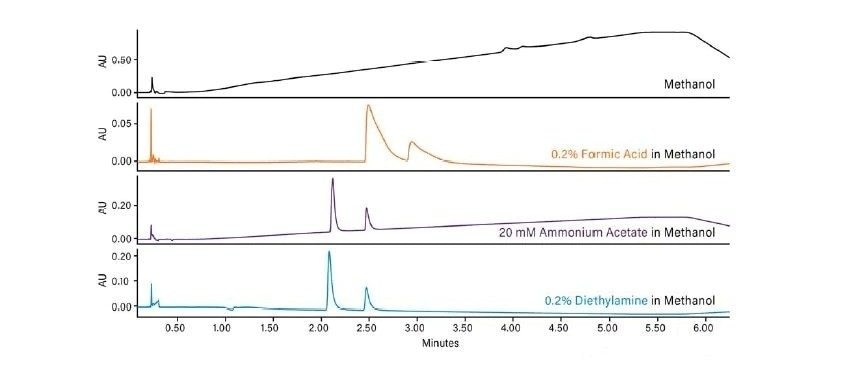
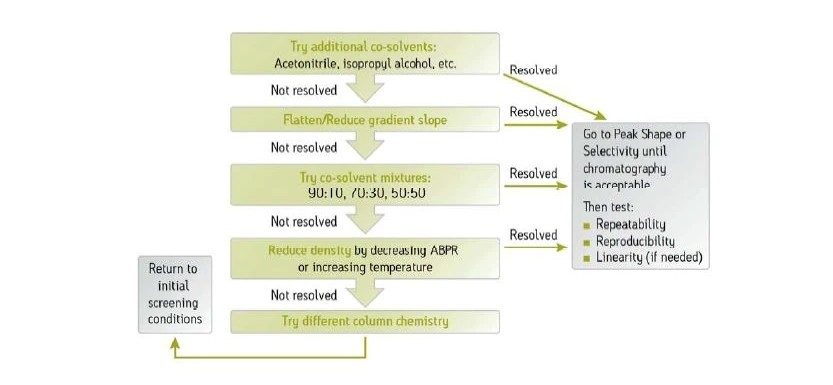
La Figure 26 illustre la façon dont la force de la phase mobile change avec différents cosolvants. Le méthanol est le cosolvant le plus fort. C'est celui qui élue les analytes le plus rapidement. L'isopropanol est plus faible que le méthanol, mais plus fort que l'acétonitrile, tandis que l'acétonitrile qui est le plus faible des trois cosolvants en CC, retient les analytes le plus longtemps. Un type relatif de comportement chromatographique identique se produit dans d'autres modes de chromatographie : les solvants plus forts diminuent la rétention et éluent les analytes plus rapidement.
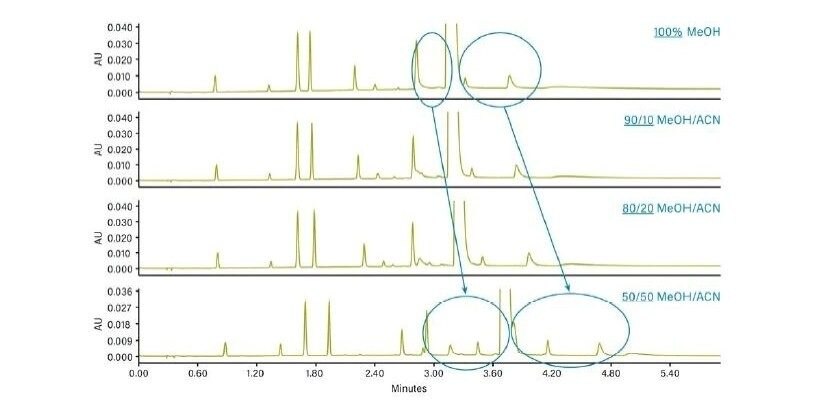
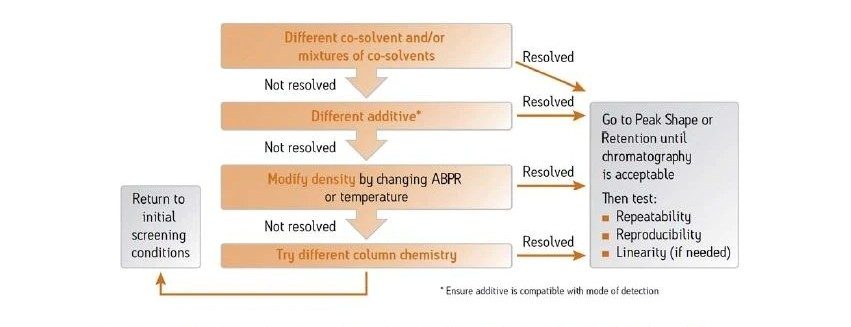
Différents cosolvants peuvent être mélangés en CC, modifiant la force du solvant et créant des différences de rétention. La Figure 27 illustre l’effet de l’ajout d’un cosolvant plus faible (acétonitrile) au méthanol, pour une séparation par gradient du métoclopramide et des impuretés associées. À mesure que la concentration en acétonitrile augmente, la concentration en méthanol et donc la force du solvant diminue, et on observe des temps de rétention plus longs. En prenant un cosolvant différent pour cette séparation, on observe de légers changements de sélectivité, une résolution améliorée et des pics qui deviennent plus fins.