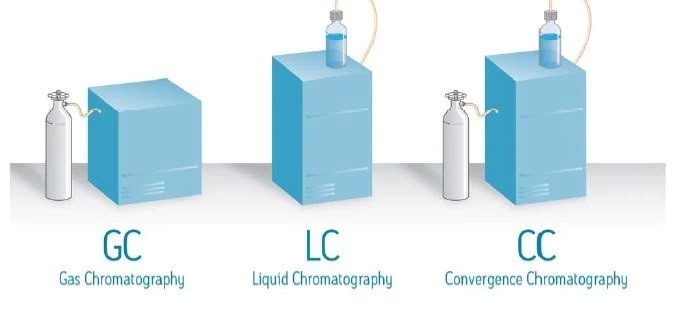
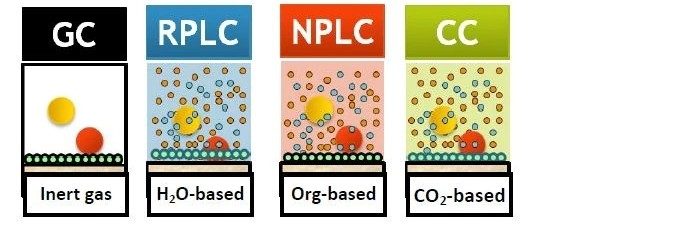
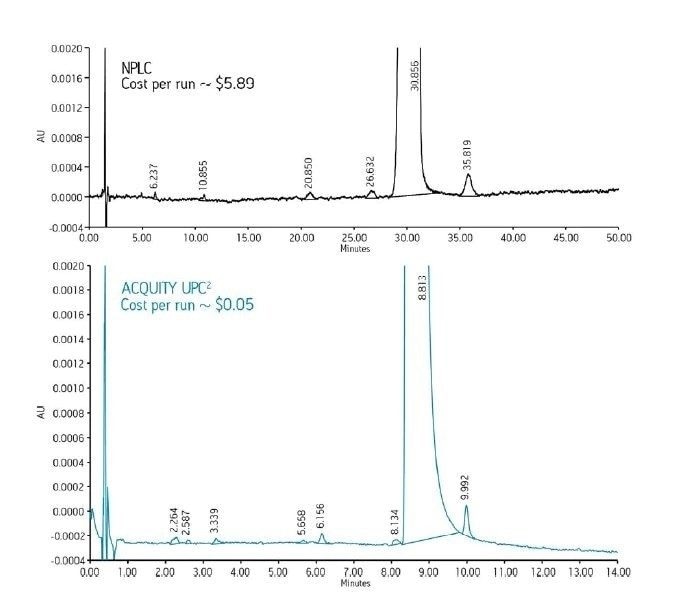
La Figure 3 présente la LC en mode RPLC et en mode NPLC. Notez que l’une des principales différences entre ces deux modes LC réside dans la composition de la phase mobile. La RPLC utilise une base d’eau, tandis que la NPLC utilise une base organique. En RPLC, la phase mobile aqueuse, conjointement à la phase stationnaire C18, modifie efficacement les interactions des analytes avec la phase stationnaire. Elle joue ainsi un rôle majeur dans la résolution des analytes des échantillons dans une grande variété de mélanges de composés. Dans le développement de méthodes RPLC, la première modification est souvent apportée à la phase mobile, et non à la phase stationnaire. En comparaison, en NPLC, la phase mobile à base d'hexane ou d'heptane joue un rôle relativement modéré et la séparation est en grande partie obtenue en modifiant la chimie de la phase stationnaire.
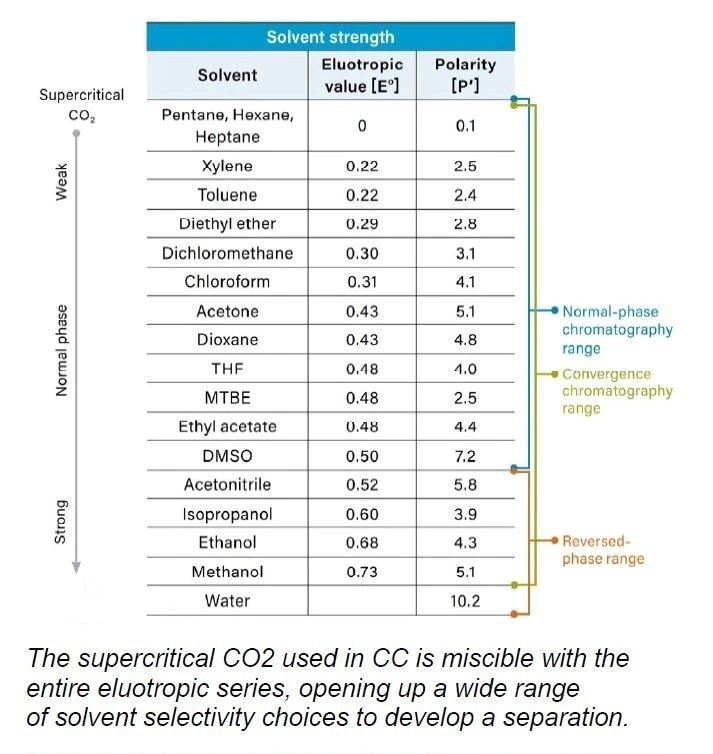
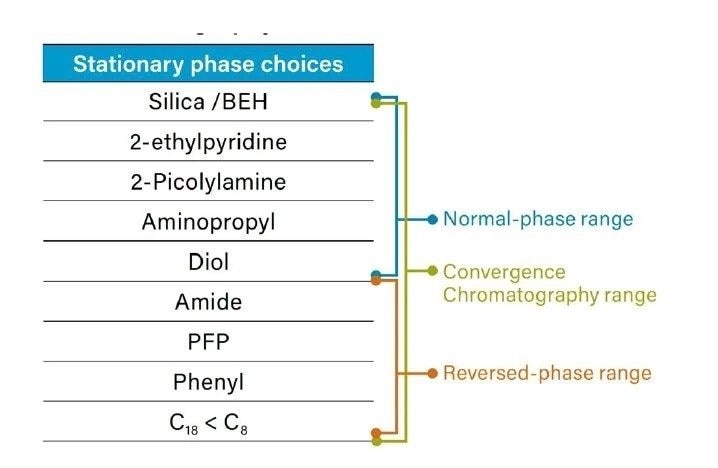
Le rôle de la phase mobile dans la CC se situe quelque part entre la RPLC et la NPLC en raison des propriétés particulières du CO2, qu’il soit supercritique ou non. Le CO2 comprimé est non polaire, comme l'heptane ou l'hexane. Sur cette base, la CC s’apparente davantage à la NPLC. Mais l'une des principales différences réside dans le fait que le CO2 est capable de se mélanger complètement aux cosolvants polaires, tels que le méthanol, l’éthanol, l’acétonitrile, etc., et peut donc être utilisé en mode gradient, contrairement à la NPLC qui est presque toujours utilisée en mode isocratique. De plus, les phases mobiles de CC sont beaucoup plus tolérantes à la présence de petites quantités d’eau que la NPLC, ce qui peut jouer un rôle important dans l’élution de l’analyte.
La section suivante présente une comparaison systématique de la CC avec la RPLC et la NPLC, en se basant sur les différences de propriétés entre leurs principaux solvants.