使用BioAccord LC-MS系统和INTACT Mass waters_connect应用程序进行RNA CQA分析
仅供研究使用,不适用于诊断。
摘要
这篇应用纪要重点介绍了分析核糖核酸(RNA)类治疗药物关键质量属性(CQA)的全新信息学功能。本研究使用由waters_connect™信息学平台控制的BioAccord™ LC-MS系统采集了离子对反相(IP-RP)液相色谱-质谱(LC-MS)数据。沃特世为不同的应用开发了多款专用软件,包括mRNA Cleaver microApp(用于生成模拟片段)、INTACT Mass应用程序(用于检测和指配RNA酶解产物)和Coverage Viewer microApp(用于酶解数据覆盖率的可视化)。实验证明此工作流程对单向导RNA (sgRNA)和信使RNA (mRNA)的CQA分析均适用,而这两种是新一代RNA治疗药物和疫苗的典型形式。
优势
- 提供用于RNA酶解片段图谱分析和核酸酶解产物质量数确认的数据分析工作流程,工作流程使用新版INTACT Mass应用程序处理数据,并辅以其他microApp工具模拟酶解物片段的生成和集展结果
- 额外提供使用INTACT Mass应用程序分析mRNA 5'端加帽效率和3'端poly(A)尾异质性的工作流程
简介
过去十年来,RNA类治疗药物和疫苗的开发势头迅猛1-3。 最近有两种基于mRNA的COVID疫苗被研发出来并成功获批,使得RNA类治疗药物成为了生物制药行业的热门话题4-6。 因此,开发监测RNA治疗药物CQA的分析方法是确保妥善控制生产工艺的当务之急。核酸治疗药物的CQA可包括活性RNA产物的序列确认、5'端加帽效率(mRNA)及结构分析、3'端poly(A)尾异质性(mRNA)分析、修饰核苷酸定位,以及纯度评估。
相关研究已经证明,LC-MS系统是表征RNA类治疗药物CQA的一种有效研究工具 7-10。但是,数据处理通常是亟待解决的瓶颈。本应用纪要介绍了一种适用于RNA CQA分析的数据处理工作流程,该工作流程用到了沃特世为waters_connect信息学平台开发的几款工具(特别是INTACT Mass、mRNA Cleaver和Coverage Viewer应用程序)。
实验
试剂和样品前处理
本文中的实验条件适用于sgRNA酶解和序列图谱分析实验。本应用纪要中关于mRNA CQA数据的实验详情和样品信息可参阅以下沃特世应用纪要:mRNA图谱分析数据(Gaye等人)、3'端poly(A)尾数据(Doneanu等人)、5'端帽数据(Ngyuen等人)13,14,18。
数据处理工作流程示例:
N,N-二异丙基乙胺(DIPEA,纯度99.5%,产品目录号387649-100ML)、1,1,1,3,3,3-六氟-2-丙醇(HFIP,纯度99%,产品目录号105228-100G)、乙醇(HPLC级,产品目录号459828-2L)和碳酸氢铵(LiChropur LC-MS Supelco试剂,产品目录号5330050050)购自Millipore Sigma(美国密苏里州圣路易斯)。乙腈(LC-MS级,产品目录号34881-1L)和甲醇(LC-MS级,产品目录号34966-1L)购自Honeywell(美国北卡罗来纳州夏洛特)。HPLC级I级去离子(DI)水使用Milli-Q系统(Millipore,美国马萨诸塞州贝德福德)净化。流动相每日现配现用。用于sgRNA酶解的无核酸酶超纯水(产品目录号J71786.AE)购自Thermo Fisher Scientific(美国马萨诸塞州沃尔瑟姆)。
100 mer sgRNA购自IDT(Integrated DNA Technologies,美国艾奥瓦州克拉尔维尔),用100 μL无核酸酶的水复溶。sgRNA在序列末端1、2、3和97、98、99这六个位置包含增强稳定性的修饰核苷酸(图1A)。修饰核苷酸包括2'-甲氧基鸟苷、2'-甲氧基尿苷和2'-甲氧基腺苷,且每种修饰都带硫代磷酸基团(图1B)。核糖核酸酶T1购自Worthington Biochemical Corporation(美国新泽西州莱克伍德)。酶解sgRNA时,将5 µL复溶的sgRNA与1 µL酶解缓冲液(100 mM碳酸氢铵)、43 µL无核酸酶超纯水和1 µL浓度为1 mg/mL的核糖核酸酶T1溶液(使用无核酸酶的水制备)混合。在QuanRecovery™ MaxPeak™ 300 µL样品瓶中制备酶解混合物,置于37 ℃下温育15 min,然后立即进行LC-MS分析。
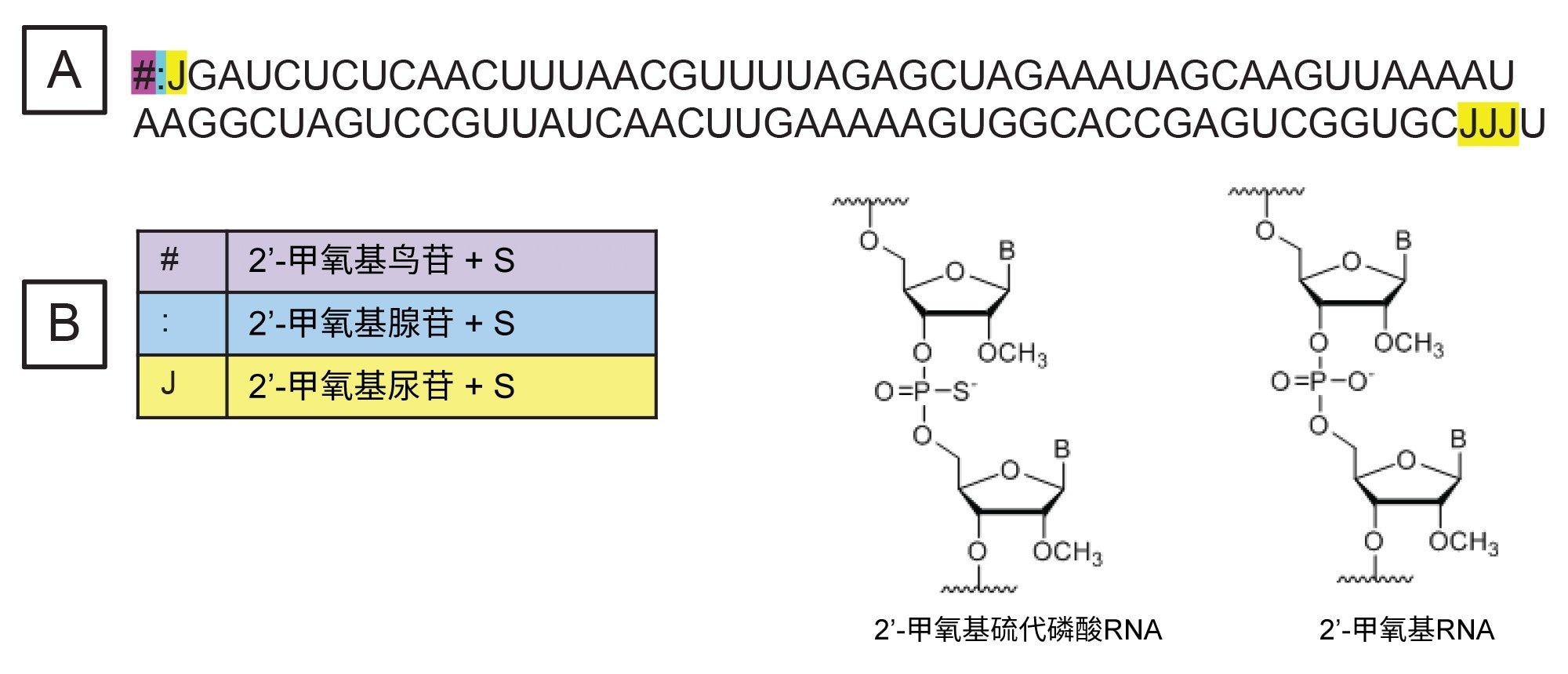 图1.待分析sgRNA的序列。“#”表示2'-甲氧基鸟苷,“:”表示2'-甲氧基腺苷,“J”表示2'-甲氧基尿苷。所有修饰核苷酸都带硫代磷酸基团。
图1.待分析sgRNA的序列。“#”表示2'-甲氧基鸟苷,“:”表示2'-甲氧基腺苷,“J”表示2'-甲氧基尿苷。所有修饰核苷酸都带硫代磷酸基团。
所有数据集都使用waters_connect信息学平台(3.2.0版)上的UNIFI™应用程序(3.6.0版)采集,并使用INTACT Mass应用程序(1.6.0版)进行处理。使用1.0.0版mRNA Cleaver和1.0.0版Coverage Viewer两款microApp。
液相色谱条件
|
LC-MS系统: |
配备ACQUITY Premier UPLC™(二元)的BioAccord LC-MS系统 |
|
保护柱: |
VanGuard FIT柱套(P/N:186007949),装有一根填充1.7 µm BEH™ C18颗粒的2.1 × 5 mm ACQUITY Premier FIT柱芯(P/N:186009459) |
|
色谱柱: |
ACQUITY Premier OST色谱柱, 1.7 µm, 130 Å, 2.1 × 150 mm(P/N:186009486) |
|
柱温: |
60 °C |
|
流速: |
300 µL/min |
|
流动相: 溶剂A: |
8 mM DIPEA(N,N-二异丙基乙胺),40 mM HFIP(1,1,1,3,3,3-六氟异丙醇),溶于去离子水中,pH 8.8 |
|
溶剂B: |
4 mM DIPEA和4 mM HFIP溶于75%乙醇 |
|
样品温度: |
6 °C |
|
样品瓶: |
QuanRecovery MaxPeak HPS样品瓶(P/N:186009186) |
|
进样体积: |
5 µL |
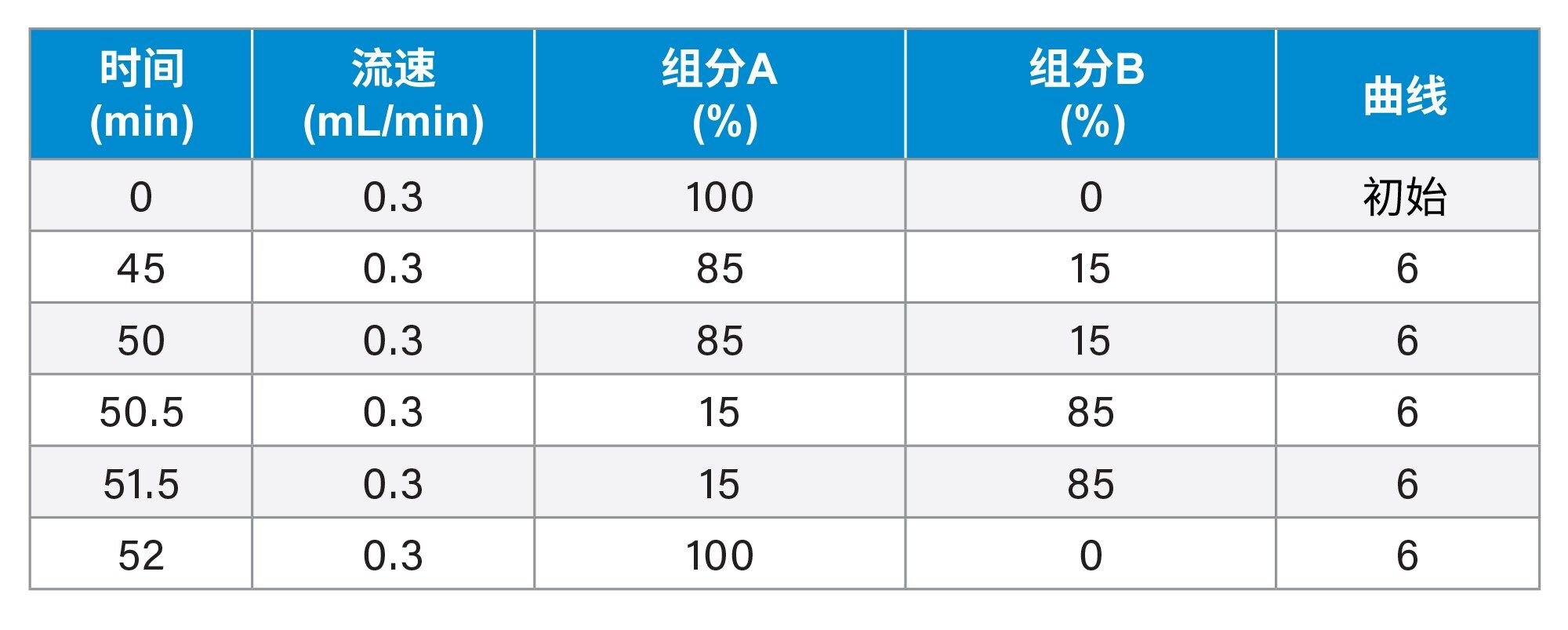
sgRNA实验的梯度表
MS条件
|
采集模式: |
全扫描 |
|
电离模式: |
ESI(-) |
|
毛细管电压: |
0.8 kV |
|
锥孔电压: |
45 V |
|
离子源温度: |
120 °C |
|
脱溶剂气温度: |
500 °C |
|
脱溶剂气体(N2)压力: |
6.5 bar |
|
TOF质量范围: |
400~5000 m/z |
|
采集速率: |
2 Hz |
|
实时校正标准液: |
waters_connect实时校正标准液(P/N:186009298) |
|
用于数据采集和处理的信息学平台: |
waters_connect v.3.2.0 |
|
数据采集: |
UNIFI应用程序3.6.0版 |
|
数据处理: |
INTACT Mass应用程序1.6.0版 |
结果与讨论
寡核苷酸图谱分析的数据处理工作流程
过去几十年来,为了满足日益增长的核酸CQA分析需求,人们开发了多种基于LC-MS的寡核苷酸图谱分析技术19-23。 寡核苷酸图谱分析与肽图分析类似,通过酶解和准确质量数测定来解析目标核酸的一级结构(序列)。寡核苷酸图谱分析常用的酶包括核糖核酸酶T1、核糖核酸酶A和MazF。本研究中的酶解使用了核糖核酸酶T1,它会在寡核苷酸3'端的每个鸟苷之后进行酶切。关于核糖核酸酶T1的作用机制,前人已经开展了大量研究,有众多文献可参考24-27。 由于只用了一种酶,我们并不期望序列覆盖率能达到100%。通常情况下,寡核苷酸图谱分析实验会并行使用多种酶,因为每种酶的特异性各不相同,产生的一组酶解片段也会有所差异。这样一来,每种酶酶解得到的片段序列就会互相重叠,进而可以提高序列覆盖率。但是,为了精简这篇应用纪要的内容,我们的实验仅使用了一种酶。
寡核苷酸图谱分析实验的另一种策略是改变酶解条件,有意使寡核苷酸部分酶解。部分酶解生成的平均核酸酶解片段更长,因此序列和质量数也更加独特。虽然这种方法可以提高覆盖率,但部分酶解可能会难以控制,导致本身就很复杂的样品额外产生(可能重叠的)m/z峰以及共流出的不完全酶解片段,复杂程度进一步增加。采集本文所示的数据时,我们的目标是完全酶解目标RNA,并为此采用了经过优化的酶解条件。
1.生成模拟酶解组分
寡核苷酸图谱分析和其他核酸CQA的数据处理工作流程如图2所示。使用mRNA Cleaver microApp生成模拟酶解片段。microApp的图形用户界面(GUI)如图3A所示。用户指南位于GUI左侧,可改善用户的软件使用体验。mRNA Cleaver microApp是一款灵活的酶解计算器兼数据库生成器,非常适合寡核苷酸图谱分析研究。
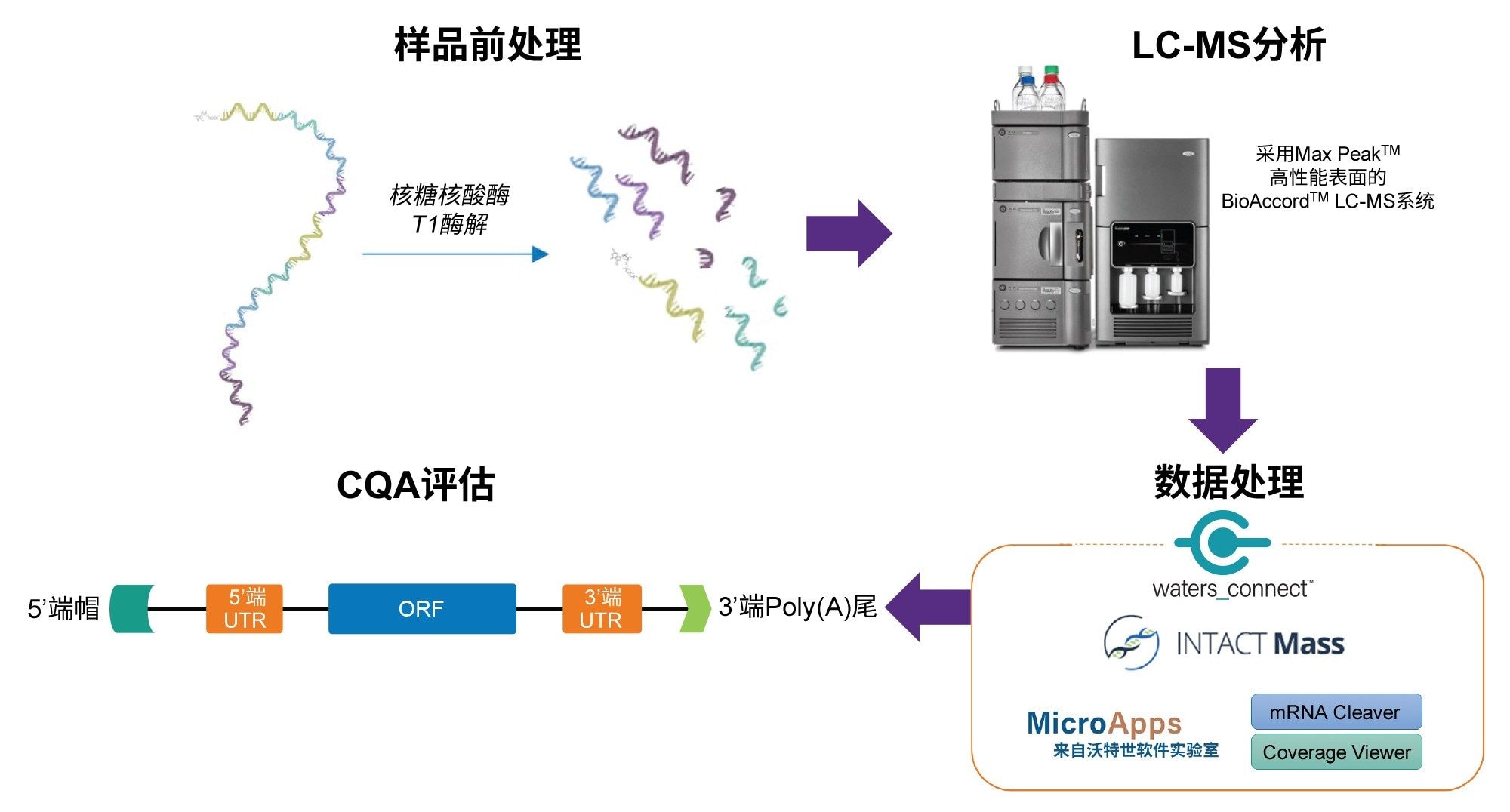 图2.RNA CQA表征工作流程示意图
图2.RNA CQA表征工作流程示意图
用户可以修改三个内置文件,根据自己的具体需求自定义设置计算器。例如,如果目标序列中含有修饰核苷酸,用户可将质量数、化学组成以及单字母或符号代码添加到“residues.csv”文件中,该文件的路径请参阅用户指南。使用修饰核苷酸的字母或符号代码可提示程序使用调整后的质量数和化学组成来计算相关的酶解产物。类似地,用户还可以修改“enzymes.csv”和“modifications.csv”文件,根据自己的实验目的添加不同的酶或mRNA特异性修饰(例如各种5'端帽或3'端poly(A)分布)。
酶解的专属设置位于Sequence(序列)字段下方(图3A),包括调整酶解产物末端、是否应生成非特异性酶解产物、要包含的漏切位点数量,以及是否考虑mRNA特异性修饰。右侧有针对生成的酶解产物列表的筛选器,它们会根据长度设置来筛选生成的酶解产物。此外,用户可以选择是否勾选Filter Duplicate Sequences(筛选重复序列)复选框。重复序列是指序列识别结果相同的酶解片段。默认情况下,mRNA Cleaver会勾选该复选框,这意味着如果有重复序列,重复序列只会列出一次,而该序列所在的其他位置会显示在另一列中。在我们的工作流程中,为了在后续数据分析步骤中使用Coverage Viewer,我们没有勾选该复选框。MS专属设置位于筛选器下方,在这里用户可以指定预期质量数离子的极性、生成的列表应采用单同位素质量数还是平均质量数、每种酶解产物生成的电荷态范围,以及要生成的m/z范围。用户单击Calculate(计算)后,将生成酶解产物列表,该列表以逗号分隔文件(.csv)形式输出。输出文件示例如图3B所示。在靶向搜索组分的处理过程中,该.csv文件将被导入INTACT Mass。
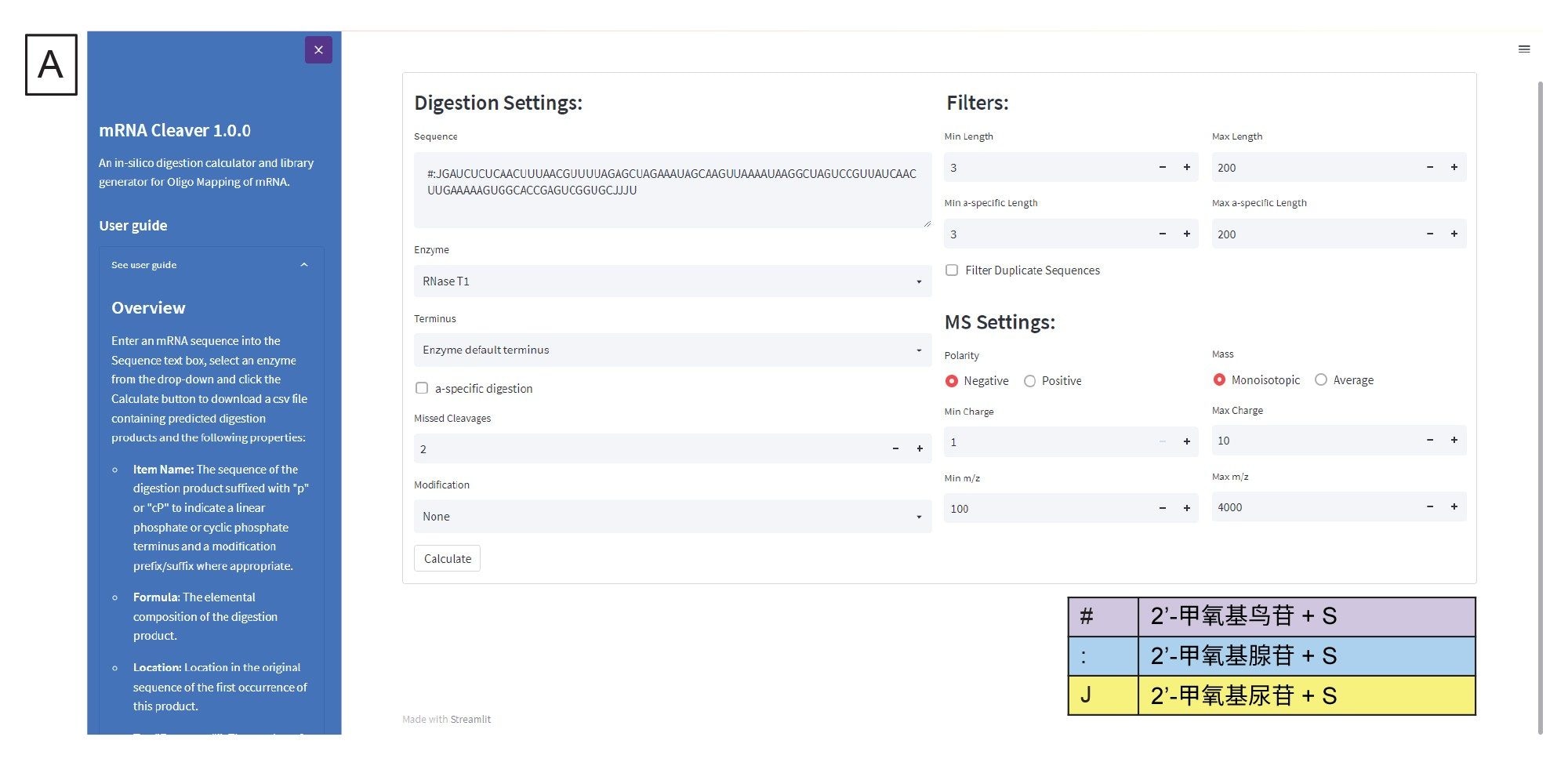 图3.(A) mRNA Cleaver microApp用户界面(GUI)
图3.(A) mRNA Cleaver microApp用户界面(GUI)
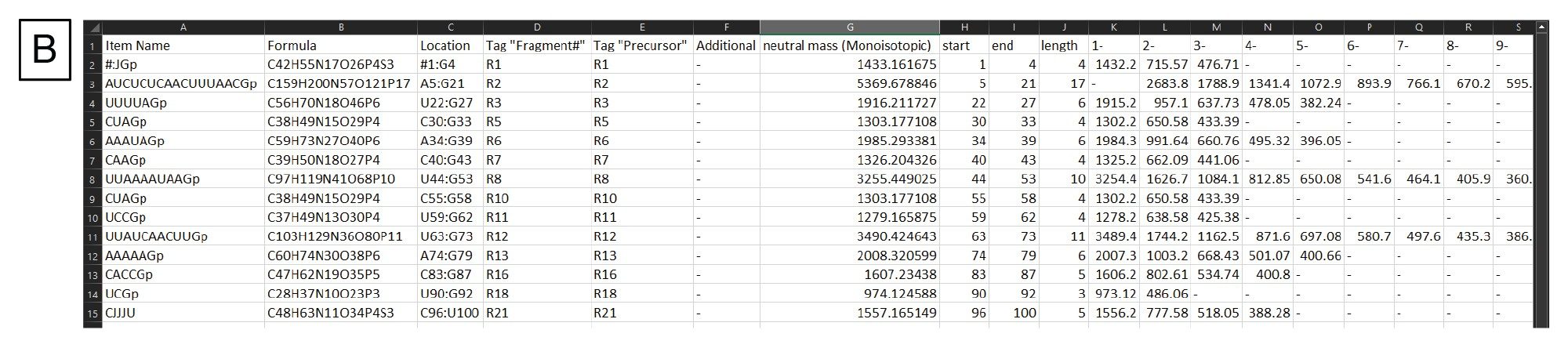 图3.(B) mRNA Cleaver输出文件示例
图3.(B) mRNA Cleaver输出文件示例
2.寡核苷酸片段质量数确认
生成模拟酶解产物列表并收集酶解样品的实验数据后,工作流程的下一步是用waters_connect中的INTACT Mass应用程序处理数据。mRNA Cleaver microApp的输出文件中有酶解产物的预期质量数及其分子ID,这些信息可直接复制到应用程序的Expected Masses(预期质量数)列。用户还可以添加修饰,包括常见加合物和序列修饰。图4A展示了分析完成后的结果界面,图4B展示了程序输出的代表性去卷积质谱图。用户可以使用表格右侧的下载按钮下载INTACT Mass结果、MS、UV和TIC表格。
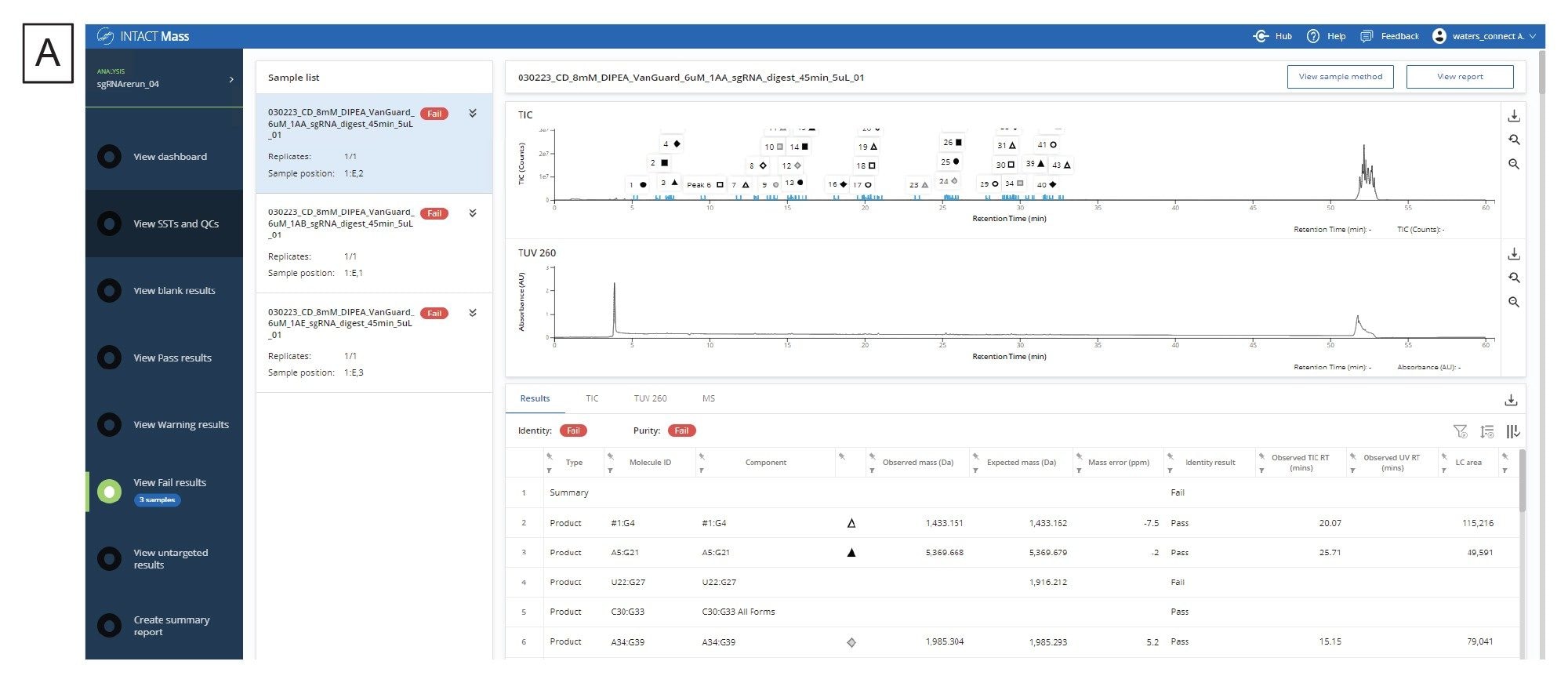 图4.(A) INTACT Mass应用程序的结果仪表板
图4.(A) INTACT Mass应用程序的结果仪表板
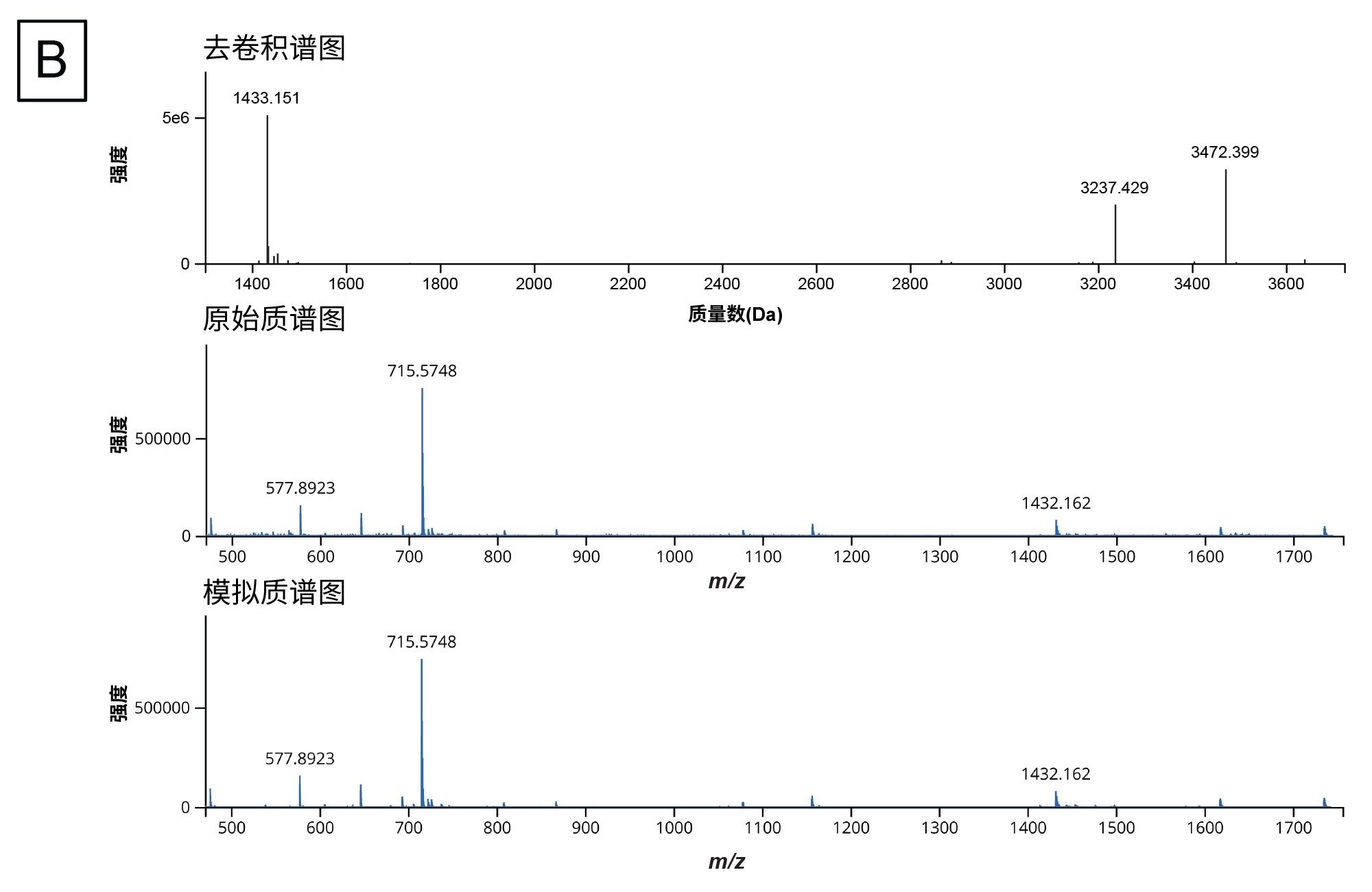 图4.(B)程序输出的去卷积质谱图示例
图4.(B)程序输出的去卷积质谱图示例
INTACT Mass应用程序生成的表格数据是报告片段图谱分析数据的关键结果,而Coverage Viewer microApp还可以比对序列将这些结果可视化,并计算序列覆盖率。Coverage Viewer microApp的GUI如图5所示。用户在Sequence(序列)字段输入目标RNA分子的序列后,即可导入工作流程第一步生成的mRNA Cleaver输出文件,填充目标酶解物片段列表(左下角)。点击左侧Import(导入)按钮可以导入INTACT Mass结果文件。序列覆盖率会以可视化形式显示在GUI右侧,程序确认目标片段之后将计算覆盖率百分比。导入INTACT Mass结果文件后,带有关键字“Pass”(通过)的酶解片段将被视为“Confirmed”(已确认)。用户也可以勾选手动指配的产物,将其视为“Confirmed”(已确认)。对应序列下方的红色区域将变为绿色,表示序列中的这个部分已根据准确质量数测定值做了确认。完成后,用户可将“Confirmed”(已确认)列被勾选的目标酶解片段部分导出为.csv文件供以后使用。此外,点击“Capture image to clipboard”(将图像捕获到剪贴板)还可以复制覆盖率图谱,以便保存到其他程序中。
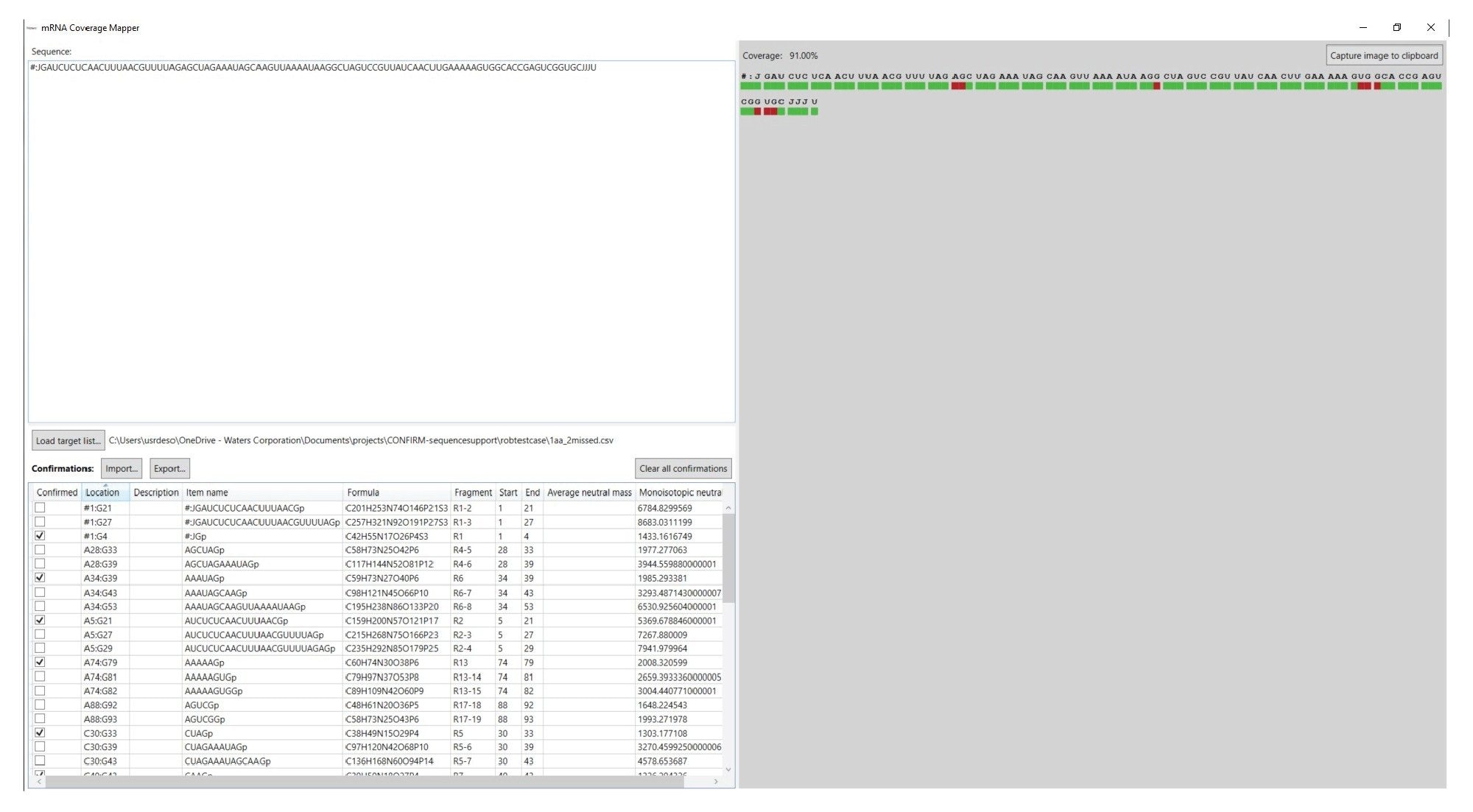 图5.Coverage Viewer microApp用户界面
图5.Coverage Viewer microApp用户界面
3.分析案例:sgRNA酶解和图谱分析
由于操作简单,基于CRISPR的基因编辑技术近年来应用越来越广泛16。该技术使用两种主要组分:向导RNA和CRISPR相关(Cas)核酸酶16。向导RNA是一种特异性RNA序列,可以识别靶DNA并将Cas核酸酶引导至正确的位点进行编辑16。Cas核酸酶是一种非特异性核酸酶,完全依靠向导RNA作用于特异性DNA位点16。最初的向导RNA由两种不同的寡核苷酸组成:与靶标互补的Crispr RNA和用作Cas核酸酶结合支架的tracr RNA16。随着技术发展,crispr RNA和tracr RNA通过名为“连接环”(linker loop)的区域融合在一起,形成单向导RNA(即sgRNA)16。合成sgRNA的一项关键CQA是序列确认,包括确认设计在分子上的特异性位点处的修饰残基。
使用我们的工作流程进行核糖核酸酶T1酶解并分析酶解产物,可以鉴定酶解产物,并在MS1水平(不需要额外的质谱碎裂)确认sgRNA序列的图谱覆盖率。首先,将sgRNA修饰核苷酸添加到residues.csv文件中,以便mRNA Cleaver通过模拟准确预测sgRNA的酶解产物。然后,调整mRNA Cleaver microApp的酶解和MS设置以反映实验条件,特别是反映实验所用的酶是核糖核酸酶T1,并且我们希望mRNA Cleaver生成带有两个漏切位点的酶解片段。即便是在完全酶解中,酶也可能漏切某些G位点,通过将漏切位点这个变量增加到2,我们考虑了这种可能性。然后程序会生成模拟酶解产物列表,并导出到.csv文件中。本研究鉴定出了两个模糊序列,即序列为CUAG的4 mer寡核苷酸。模糊序列是指同分异构和/或同量异位的酶解片段,这意味着这些序列的m/z峰和电荷态峰簇会互相重叠。如果模糊序列覆盖了RNA中对于分析至关重要的部分,可能需要进一步开展实验(例如在气相中碎裂)来确认序列鉴定结果。否则,其他19种酶解片段都有唯一的预期质量数和序列鉴定结果,则通过质谱图应该可以判断。这个假设的依据是BioAccord生成的预期质量数的精度,以及mRNA Cleaver microApp为每个酶解片段生成的模拟m/z范围。
我们按照实验方法中的说明做了体外酶解,并使用waters_connect在BioAccord LC-MS系统上收集了酶解后sgRNA和未酶解的完整sgRNA的数据。图6A展示了完整sgRNA及其酶解之后的总离子流色谱图。酶解片段在梯度的5~40 min之间洗脱。完整sgRNA在35.6 min洗脱,而酶解样品迹线在该时间处没有对应的峰,这表明sgRNA已完全酶解。
我们将LC-MS数据和模拟酶解组分列表都提交给了INTACT Mass应用程序,以便通过质量数匹配(匹配容差为10 ppm)来指配各种酶解组分。然后将INTACT Mass结果导入Coverage Viewer microApp,对序列覆盖率进行可视化处理,如图6B所示。MS1序列覆盖率高达91%。在本例中,我们未通过碰撞诱导碎裂来确认序列鉴定结果,因为模糊序列的鉴定结果都是相同的,没有必要做这一步操作。但是,如果存在许多模糊序列,或者目标模糊序列具有不同的序列鉴定结果,除非这些模糊序列的鉴定结果可通过碎裂得到确认,否则分析人员在计算序列覆盖率时可能会想要排除它们28。为达到100%序列覆盖率,可以使用酶切特异性不同于核糖核酸酶T1的另一种内切核糖核酸酶(例如MasF或核糖核酸酶A)来覆盖sgRNA序列中未鉴定的部分。
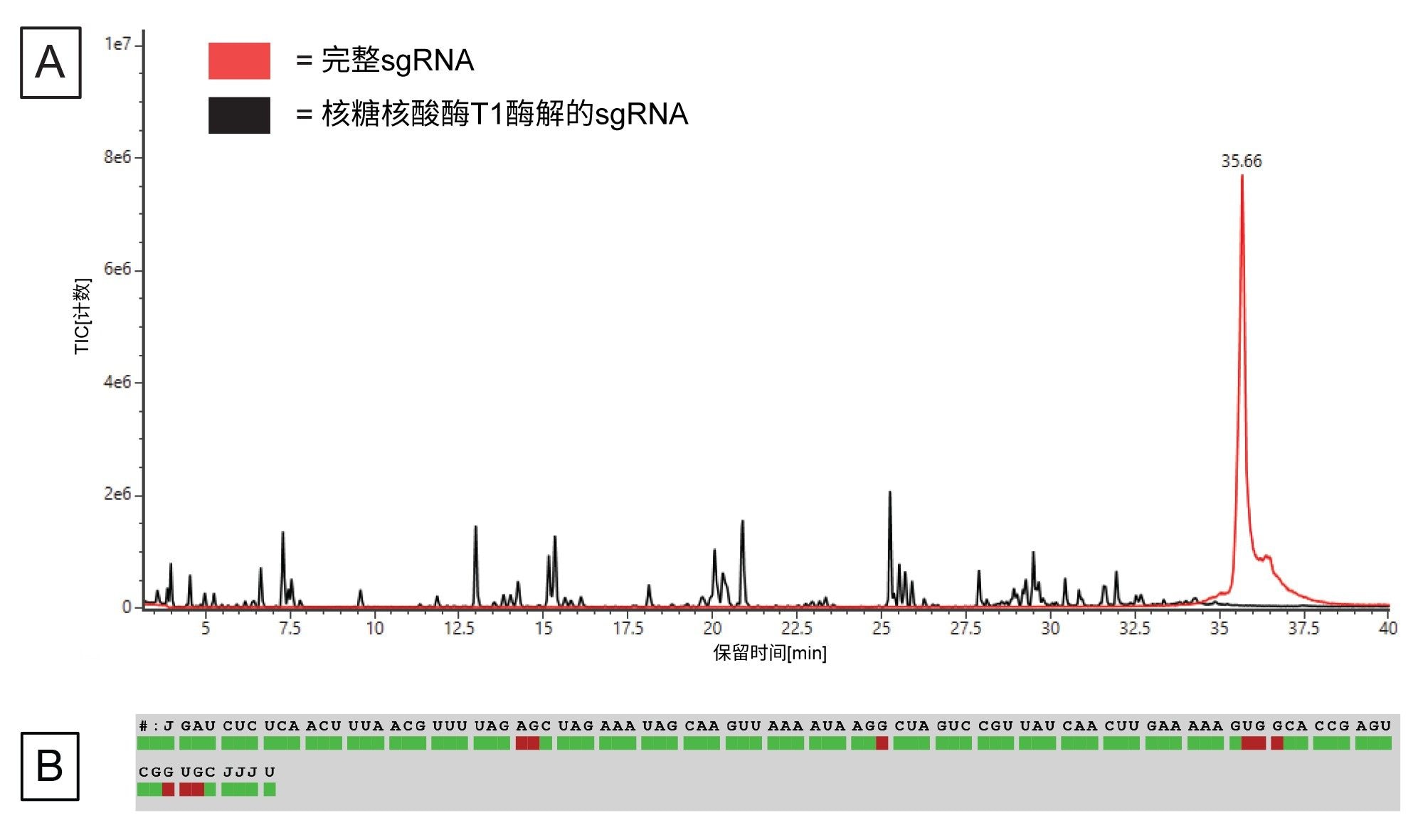 图6.(A)酶解后的sgRNA(黑色迹线)和未酶解的完整sgRNA(红色迹线)的TIC,(B) Coverage Viewer microApp分析LC-MS数据后得出的序列覆盖率。
图6.(A)酶解后的sgRNA(黑色迹线)和未酶解的完整sgRNA(红色迹线)的TIC,(B) Coverage Viewer microApp分析LC-MS数据后得出的序列覆盖率。
mRNA酶解和图谱分析
此工作流程还可应用于大分子RNA(例如信使RNA (mRNA))。然而,在MS1水平对大分子RNA进行图谱分析是一项高难度的任务,因为分析时会额外产生许多模糊序列和共流出物质,导致数据复杂程度呈指数级增长。Gaye等人之前发表的一篇应用纪要介绍了一种使用IP-RP LC-MS系统和waters_connect平台执行mRNA序列图谱分析的优化方法18。本研究使用更新后的工作流程重新处理了数据。
图7A包含Gaye等人得到的核糖核酸酶T1 mRNA酶解物的TIC色谱图以及Coverage Viewer得出的结果18。插图是从8.58 min处洗脱峰提取的质谱图(XIC)。这幅代表性的XIC凸显出数据的复杂性,因为该峰中有许多物质共流出。按照工作流程,我们先使用mRNA Cleaver microApp生成了两份模拟酶解产物列表:一份包含模糊序列,另一份剔除模糊序列。接着将这些列表以及采集到的LC-MS数据提交至INTACT Mass应用程序进行数据处理。然后,将INTACT Mass结果导入Coverage Viewer microApp。
MS1水平的酶解片段图谱表明,特有序列覆盖率为38.87%,总体序列覆盖率为75.18%(包括模糊序列)。这些图谱分别如图7B和7C所示。 虽然并行使用另一种内切核糖核酸酶还可以进一步提高序列覆盖率,但数据复杂程度增加(共流出物质、模糊序列等)将始终是数据解析面临的一大挑战。处理更复杂样品所需的时间与待分析的峰数量成正比,可能成为分析通量的限制因素。
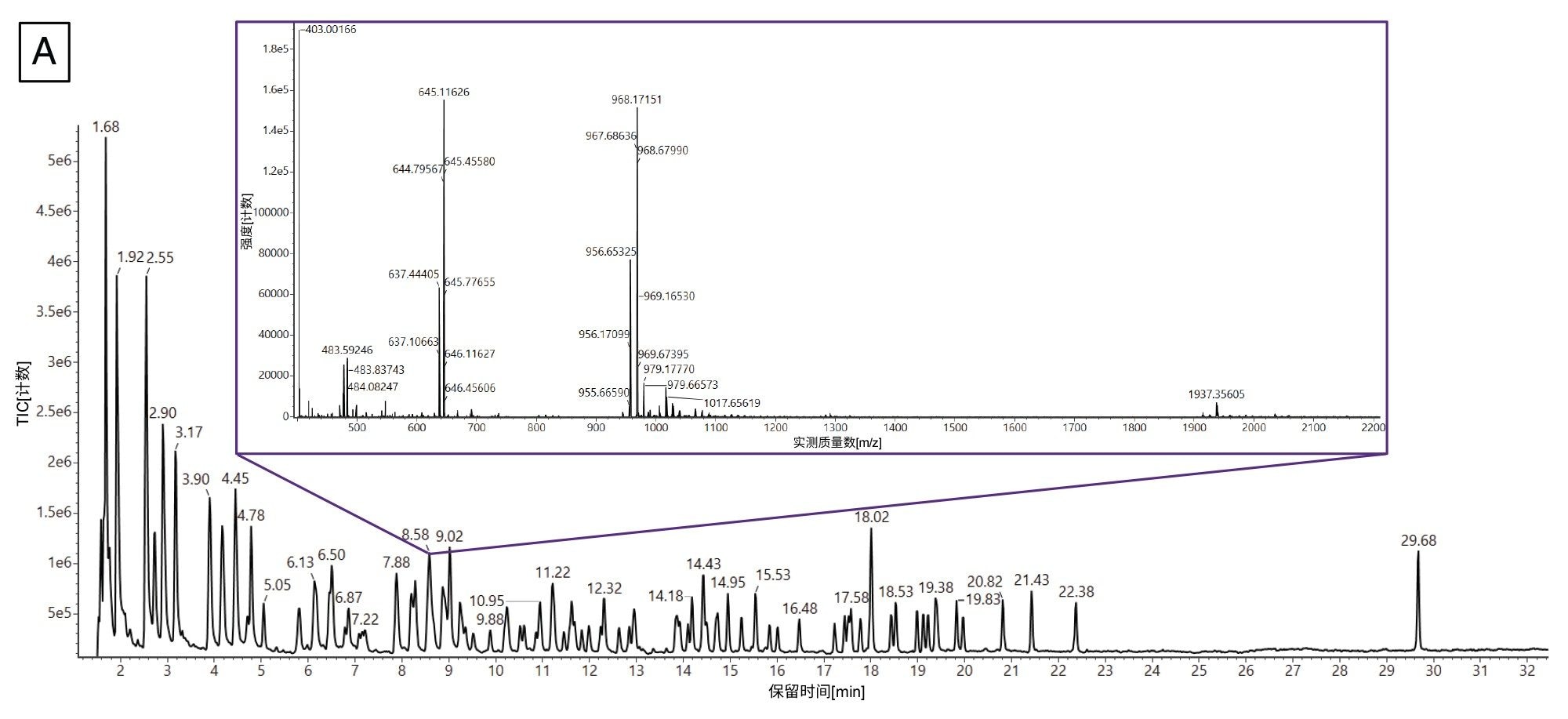 图7.(A) Gaye等人得到的mRNA酶解物的TIC(18)。插图为8.58 min处洗脱峰的代表性XIC,其中包含几种共流出的寡核苷酸。
图7.(A) Gaye等人得到的mRNA酶解物的TIC(18)。插图为8.58 min处洗脱峰的代表性XIC,其中包含几种共流出的寡核苷酸。
 图7.(B) Coverage Viewer microApp处理MS1数据得出的特有序列覆盖率(不含模糊序列)
图7.(B) Coverage Viewer microApp处理MS1数据得出的特有序列覆盖率(不含模糊序列)
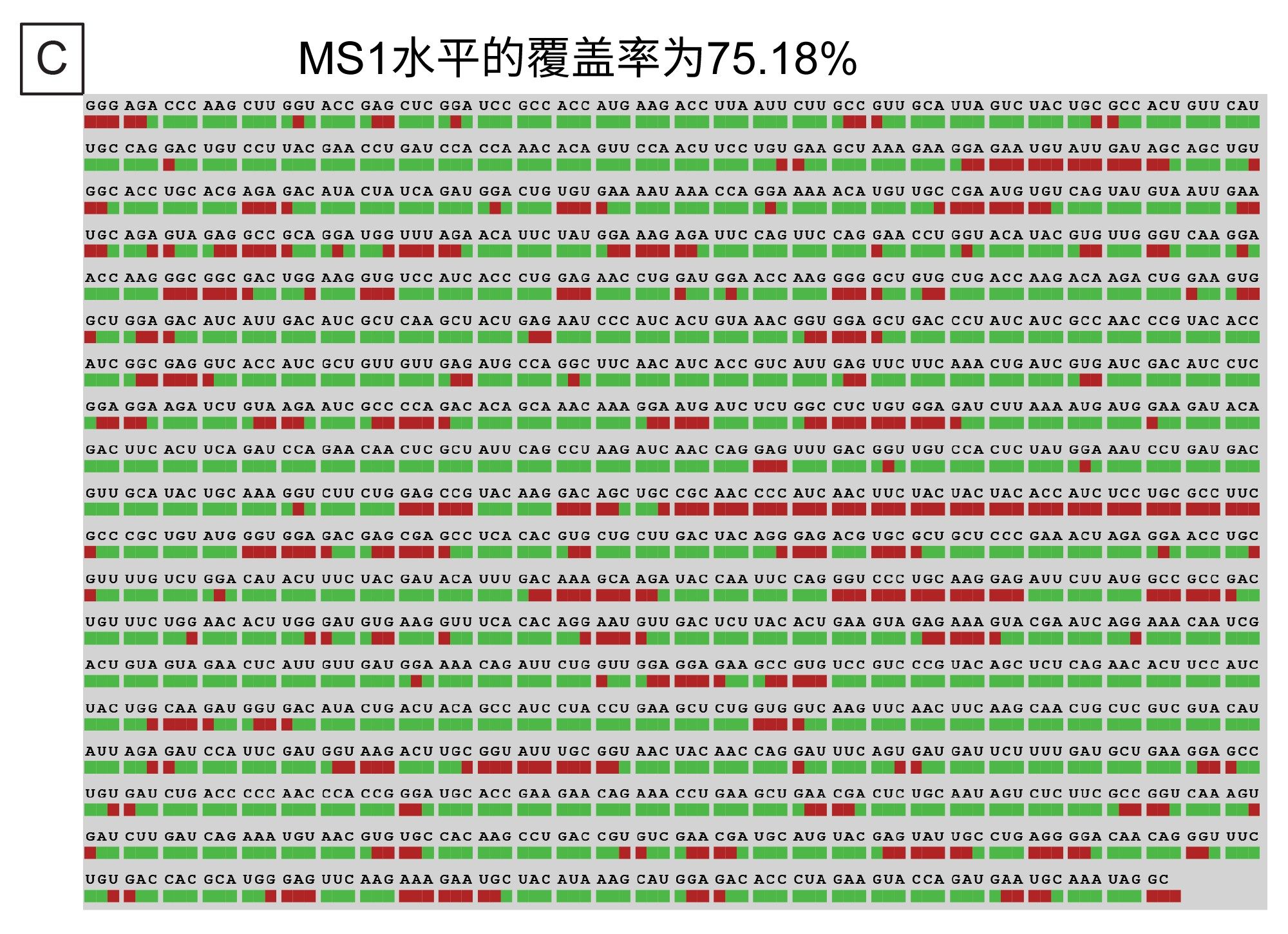 图7.(C) Coverage Viewer microApp处理MS1数据得出的总体序列覆盖率
图7.(C) Coverage Viewer microApp处理MS1数据得出的总体序列覆盖率
如果使用BioAccord的图谱分析实验还收集了数据非依赖型(MSE)碎裂数据,则可以使用CONFIRM Sequence应用程序进一步解析模糊片段。Gaye等人做了此类分析,示例数据如图8所示18。 相关研究正在为这个附加分析步骤开发自动化工作流程。
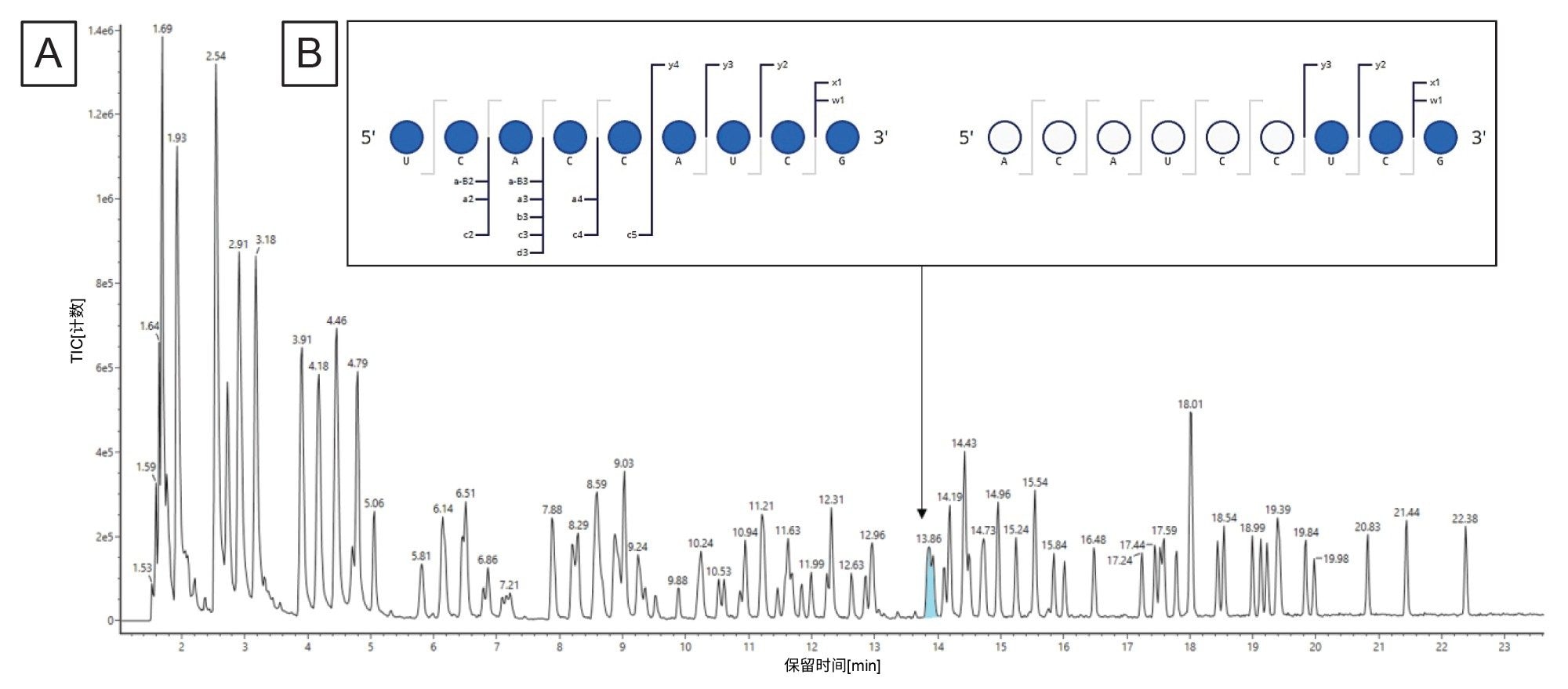 图8.(A) Gaye等人预测了623–631(ACAUCCUCGp)和551–559 (UCACCAUCGp)这两个位置的酶解片段组分,并将它们指配给了得到的TIC中的同一个RT峰。无法通过完整质量数信息确定正确的指配结果。(B)同一进样的MSE数据可用于阐明此指配结果的正确序列。使用waters_connect CONFIRM Sequence应用程序预测每个序列的高能量碎片离子,并通过定制算法与积分原始数据的同位素簇相匹配。该软件在点图上显示了已确认的碎片离子,可用于快速评估序列覆盖率。
图8.(A) Gaye等人预测了623–631(ACAUCCUCGp)和551–559 (UCACCAUCGp)这两个位置的酶解片段组分,并将它们指配给了得到的TIC中的同一个RT峰。无法通过完整质量数信息确定正确的指配结果。(B)同一进样的MSE数据可用于阐明此指配结果的正确序列。使用waters_connect CONFIRM Sequence应用程序预测每个序列的高能量碎片离子,并通过定制算法与积分原始数据的同位素簇相匹配。该软件在点图上显示了已确认的碎片离子,可用于快速评估序列覆盖率。
测定5'端加帽效率
mRNA特有的一项CQA是评估5'端加帽效率和结构。mRNA分子的5'端帽对于多种生物学功能至关重要,包括调节核输出、防止mRNA被核酸外切酶降解、辅助翻译过程,以及辅助5'端近端内含子切除14。在之前的一篇应用纪要中,Nguyen等人提出了一种使用BioAccord LC-MS系统快速分析合成mRNA端帽结构的方法(用时不到5 min)14。
使用我们的工作流程,我们根据去卷积质谱图(如图9A所示)成功检测到5'端帽杂质并指配了结构。分析这些数据时,我们应用了自定义电荷去卷积,这是INTACT Mass应用程序(1.6.0及更高版本)的一项新功能。自动去卷积功能采用经过优化的设置执行峰检测和去卷积处理,适用于简化复杂程度各异的多种样品的常规分析。但是,这些数据包含动态范围很宽的分析物,其中有我们希望稳定鉴定的几种接近分析检测下限的低强度杂质,因此我们发现自动去卷积算法设置并不适合这种情况。自定义去卷积功能便于科学家优化谱图去卷积设置,让谱图更好地代表分子上的全部5'端帽结构。
我们对这些数据应用了BayesSpray电荷去卷积算法,并专门为数据定制了保留时间范围、输入m/z范围、输出预期质量范围和电荷范围。5'端帽分析的INTACT Mass数据如图9B所示。我们在1:10稀释样品中成功鉴定出了所有杂质(ppG、pppG、GpppG和m7GpppG(端帽-0)),还在按Ngyuen等人的方法制备的1:100稀释样品中成功鉴定出了前面提到的三种杂质(pppG除外)14。
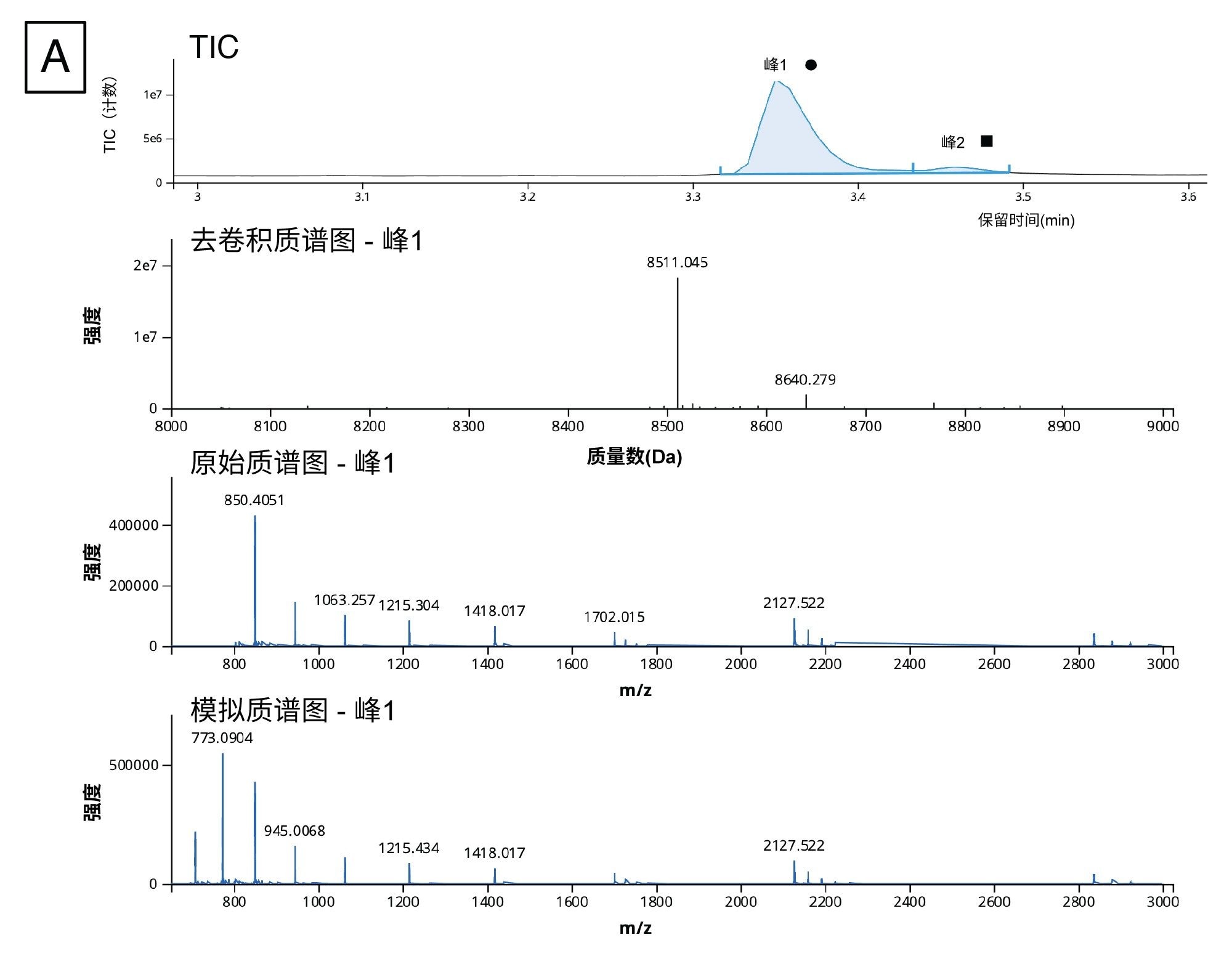 图9.(A) TIC及其3.37 min处的峰(圆圈)的去卷积质谱图、原始谱图和模拟谱图
图9.(A) TIC及其3.37 min处的峰(圆圈)的去卷积质谱图、原始谱图和模拟谱图
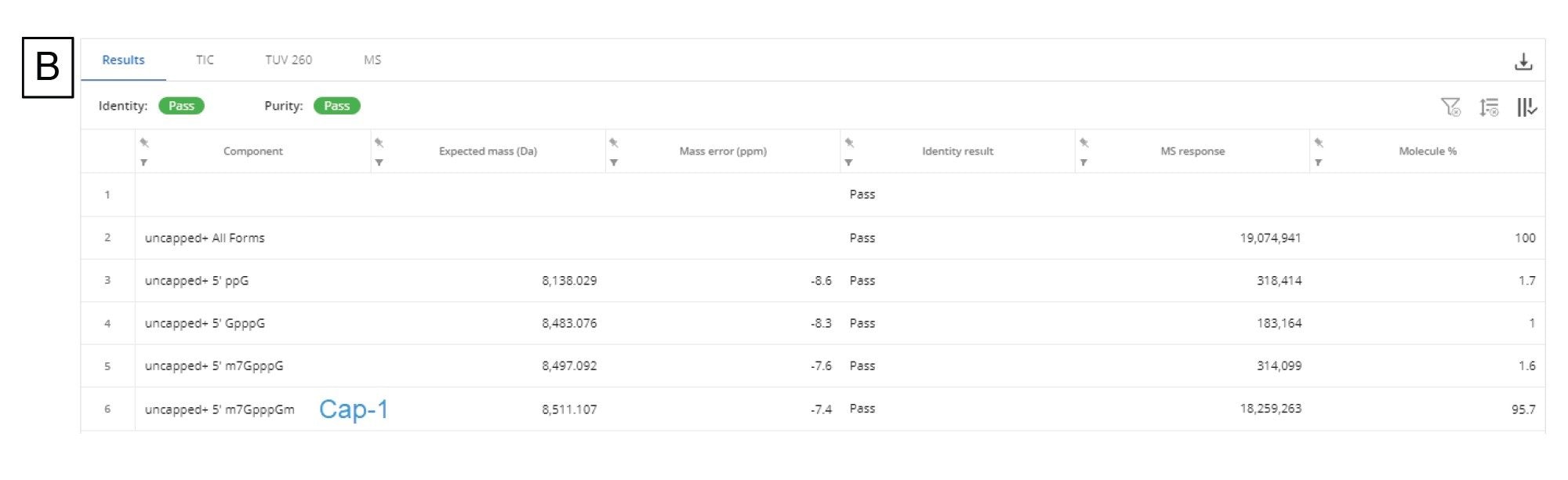 图9.(B)端帽-1片段及其产物相关杂质片段的INTACT Mass应用程序结果表
图9.(B)端帽-1片段及其产物相关杂质片段的INTACT Mass应用程序结果表
翻译效率很大程度上依赖于mRNA的5'端,因此,正确加帽的mRNA产物所占的百分比是mRNA类治疗药物生产过程要密切监测的CQA项目之一。Nguyen等人开发的方法配合使用自定义去卷积设置的数据分析工作流程可以提供足够的灵敏度和稳健性,用于常规监测不良加帽过程产生的杂质14。
可靠地测定3'端poly(A)尾长度和分布
mRNA特有的另一项CQA是确定3'端poly(A)尾的长度。3'端poly(A)尾的长度与mRNA稳定性直接相关17。 因此,测定长度(确切地说是测定平均长度和大小分布)有助于我们深入了解多聚腺甘酸化过程。Doneanu等人开发出了一种相对快速的LC-MS分析方法(运行时间15 min),用于分析萤火虫荧光素酶(FLuc) mRNA经核糖核酸酶T1酶解后产生的3'端poly(A)尾寡核苷酸13。如图10A所示,3'端poly(A)尾峰在TIC中晚洗脱,分离良好且丰度较高。我们将相应的ESI-MS合并谱图(包含几种共流出的3'端poly(A)尾分析物)提交给工作流程,使用INTACT Mass应用程序中的MaxEnt1电荷去卷积算法做了去卷积处理。处理时调整了自定义电荷去卷积参数(例如输入m/z范围、输出质量范围和保留时间范围),生成了如图10A插图所示的去卷积质谱图。该谱图表明3'端poly(A)尾存在广泛的异质性,寡核苷酸大小在122~132 mer之间,彼此间相差的特征性质量数(329.2059 Da)为单磷酸腺苷残基的质量数。INTACT Mass结果如图10B所示,检测到的所有寡核苷酸组分的ESI-MS信号及其相应的强度都汇总在图10C中。该图被用于计算FLuc mRNA 3'端poly(A)尾的重均质量数。测得的平均质量数对应的长度为126.5 mer,通过正交SEC测定确认了此测定结果15。INTACT Mass处理后,我们测定了11种3'端poly(A)尾异构体,其典型质量精度均优于45 ppm(图10B)。
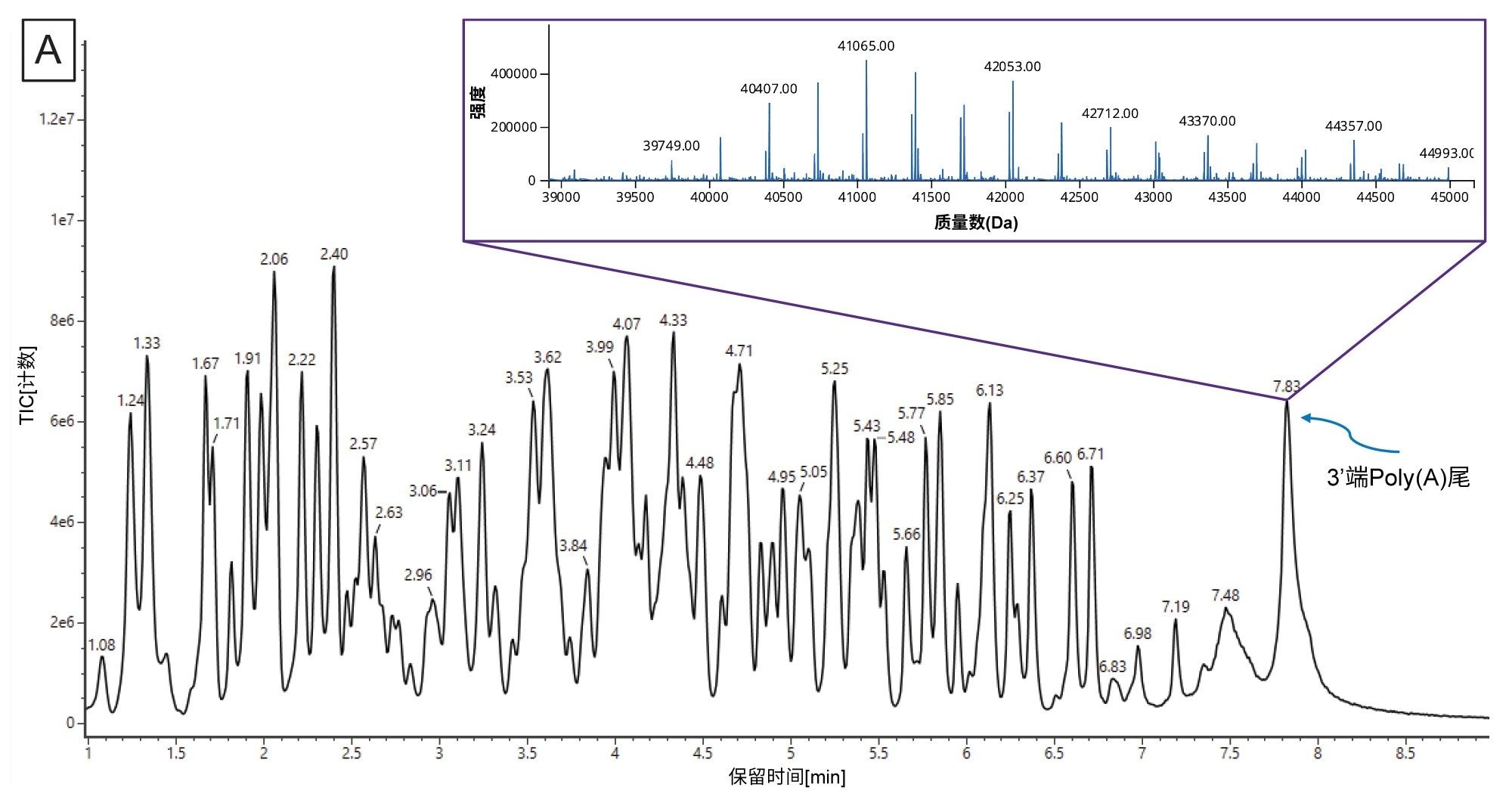 图10.(A) FLuc mRNA经核糖核酸酶T1酶解后得到的3'端poly (A)尾寡核苷酸混合物的TIC色谱图。插图展示了INTACT Mass应用程序的MaxEnt1电荷去卷积算法得出的结果。
图10.(A) FLuc mRNA经核糖核酸酶T1酶解后得到的3'端poly (A)尾寡核苷酸混合物的TIC色谱图。插图展示了INTACT Mass应用程序的MaxEnt1电荷去卷积算法得出的结果。
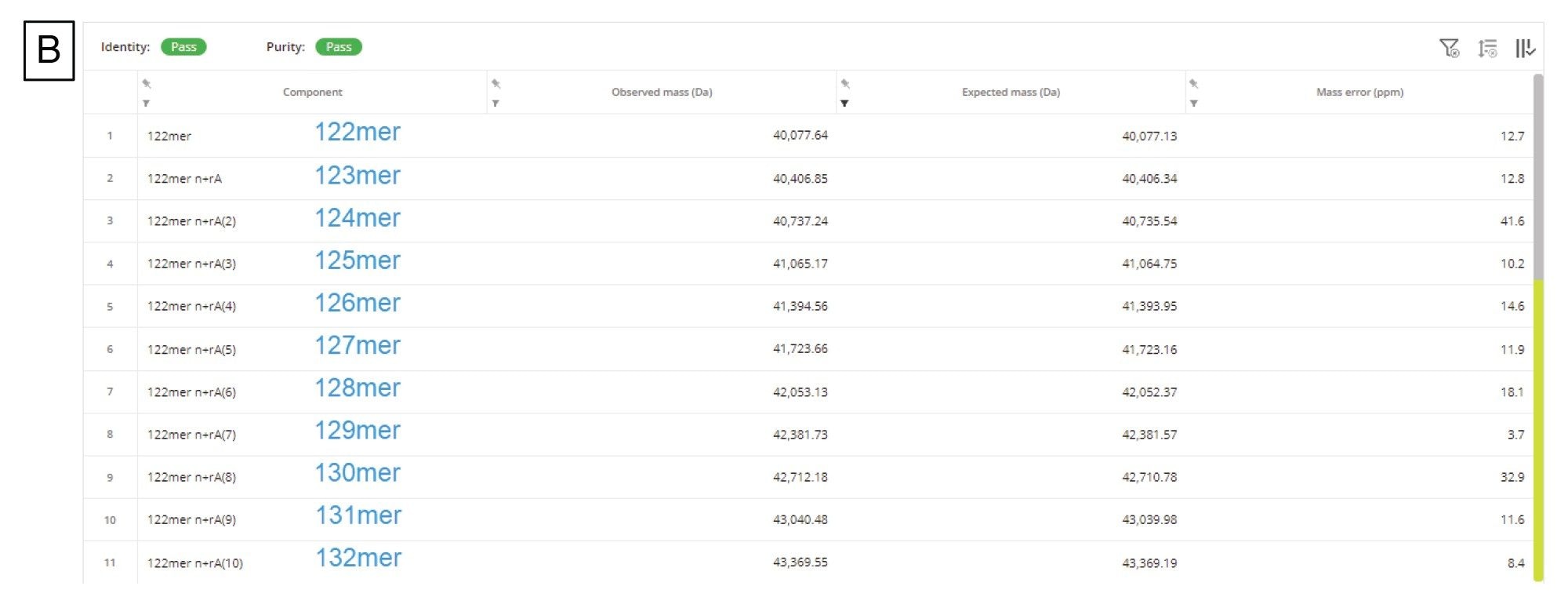 图10.(B) INTACT Mass应用程序结果表,由表可知3'端poly(A)尾特征呈广泛分散性,在检测到的第一个poly(A)尾异构体(122 mer)的基础上,最多增加了10个腺苷的质量数。在INTACT Mass中使用自定义去卷积参数处理数据后,成功鉴定出11种寡核苷酸(长度从122到132 mer不等),且质量精度均优于45 ppm。
图10.(B) INTACT Mass应用程序结果表,由表可知3'端poly(A)尾特征呈广泛分散性,在检测到的第一个poly(A)尾异构体(122 mer)的基础上,最多增加了10个腺苷的质量数。在INTACT Mass中使用自定义去卷积参数处理数据后,成功鉴定出11种寡核苷酸(长度从122到132 mer不等),且质量精度均优于45 ppm。
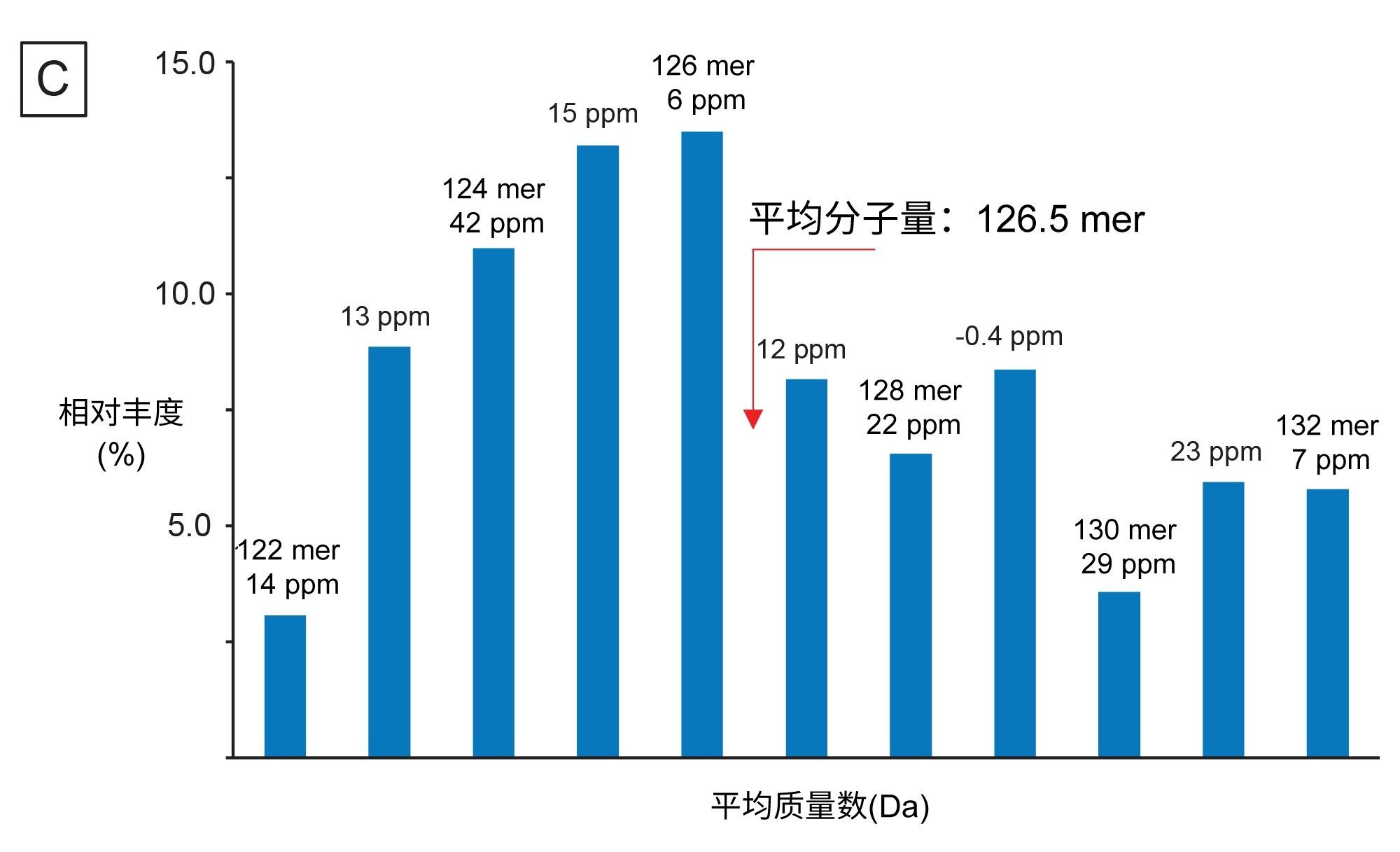 图10.(C) ESI-MS谱图强度相对于3'端poly(A)尾长度所做的分布图,根据该图可确定平均poly(A)尾长度和分散性。
图10.(C) ESI-MS谱图强度相对于3'端poly(A)尾长度所做的分布图,根据该图可确定平均poly(A)尾长度和分散性。
3'端poly(A)尾分散性和平均质量数测定值都可以衡量工艺稳健性和潜在临床效果,是mRNA分子类治疗药物的重要CQA。综上,实验证明Doneanu等人开发的LC-MS分析方法配合我们使用INTACT Mass应用程序的数据分析工作流程可以有效分析mRNA类预防用药/治疗药物酶解后产生的3'端poly(A)尾。
结论
- 采用在BioAccord LC-MS系统和waters_connect信息学平台上开发的工作流程,我们可通过序列图谱确认(寡核苷酸、sgRNA和mRNA)、5'端加帽效率分析(mRNA)和3'端poly(A)尾分析(mRNA)等方法开展长链寡核苷酸和现代RNA治疗药物的CQA分析。
- 为了支持这些工作流程,INTACT Mass应用程序增加了强化的信息学功能。全新的自定义去卷积功能可实现自动化的常规数据处理,即使样品信号强度较低或者目标分析物动态范围很宽也能从容应对。
- 沃特世专为RNA数据分析开发的其他microApp还可以补充INTACT Mass应用程序的这些强化功能:
- mRNA Cleaver可应用用户自定义的残基、酶和mRNA修饰生成模拟酶解产物列表
- Coverage Viewer可以轻松完成酶解片段图谱序列覆盖率的计算和可视化
参考资料
- Zhu, Y., Zhu, L., Wang, X. & Jin, H. RNA-based Therapeutics: An overview and prospectus.Cell Death & Disease 13, (2022).
- Crooke ST, Witztum JL, Bennett CF, Baker BF.RNA-Targeted Therapeutics.Cell Metab.2018 Apr 3;27(4):714-739.doi: 10.1016/j.cmet.2018.03.004. Erratum in: Cell Metab.2019 Feb 5;29(2):501.PMID: 2961764
- Damase, T. R. et al.The Limitless Future of RNA therapeutics.Frontiers in Bioengineering and Biotechnology 9, (2021).
- Xu, S.; Yang, K.; Li, R.; Zhang, L., mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. International Journal of Molecular Sciences 2020, 21 (18), 6582
- Verbeke, R., Lentacker, I., De Smedt, S. C. &; Dewitte, H. The dawn of mRNA vaccines: The COVID-19 case. Journal of Controlled Release 333, 511–520 (2021).
- Jackson, N. A., Kester, K. E., Casimiro, D., Gurunathan, S. &; DeRosa, F. The Promise of mRNA vaccines: A biotech and industrial perspective.npj Vaccines 5, (2020).
- Goyon, A. et al.Full sequencing of CRISPR/Cas9 single guide RNA (sgRNA) via parallel ribonuclease digestions and hydrophilic interaction liquid chromatography–high-resolution mass spectrometry analysis.Analytical Chemistry 93, 14792–14801 (2021).
- Ivleva, V. B., Yu, Y.-Q. &; Gilar, M. Ultra-performance liquid chromatography/tandem mass spectrometry (UPLC/MS/MS) and UPLC/MSe analysis of RNA oligonucleotides.Rapid Communications in Mass Spectrometry 24, 2631–2640 (2010).
- Jiang, T. et al.Oligonucleotide sequence mapping of large therapeutic mRNAs via parallel ribonuclease digestions and LC-MS/MS.Analytical Chemistry 91, 8500–8506 (2019).
- Ross, R., Cao, X. & Limbach, P. Mapping post‐transcriptional modifications onto transfer ribonucleic acid sequences by liquid chromatography tandem mass spectrometry.Biomolecules 7, 21 (2017).
- Shion, H., Boyce, P., Berger, S. & Yu, Y. Q. INTACT Mass™ - 一款用于生物治疗药物快速质量数确认和纯度评估的通用waters_connect™应用程序.沃特世应用纪要, 720007547ZH, 2023年2月.
- Jackson, N. A., Kester, K. E., Casimiro, D., Gurunathan, S. & DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective.npj Vaccines 5, (2020).
- Doneanu, C., Fredette, J. & Yu, Y. Q. Ion-Pairing Reversed Phase LC-MS Analysis of Poly(A) Heterogeneity Using the BioAccord LC-MS System, Waters Application Note, 720007925, 2023.
- Nguyen, J. M. et al. 使用离子对反相液相色谱法和BioAccord LC-MS系统快速分析合成的mRNA帽状结构.沃特世应用纪要, 720007329ZH, 2023年10月.
- Gilar, M. 体积排阻色谱法分析mRNA的Poly(A)尾.沃特世应用纪要, 720007853ZH, 2023年10月.
- Hu, L., Li, Y., Wang, J., Zhao, Y. & Wang, Y. Controlling CRISPR-Cas9 by guide RNA engineering.WIREs RNA 14, (2022).
- Pelletier, J.; Sonenberg, N. The Organizing Principles of Eukaryotic Ribosome Recruitment.Annu.Rev.Biochem.2019, 88, 307–335
- Gaye, M. M. et al. 使用离子对液相色谱和质谱法进行合成mRNA寡核苷酸映射.沃特世应用纪要, 720007669ZH, 2023年10月.
- Fekete, S et al.Challenges and emerging trends in liquid chromatography-based analyses of mRNA pharmaceuticals.Journal of Pharmaceutical and Biomedical Analysis.224 (2022).
- Fountain, K. J., Gilar, M. & Gebler, J. C. Analysis of native and chemically modifed oligonucleotides by tandem ion-pair reversed phase high-performance liquid chromatography/electrospray ionization mass spectrometry.Rapid Commun.Mass Spectrom. 17, 646–653. https://doi.org/10.1002/rcm.959 (2003).
- Zhang, G., Lin, J., Srinivasan, K., Kavetskaia, O. & Duncan, J. N. Strategies for bioanalysis of an oligonucleotide class macromolecule from rat plasma using liquid chromatography−tandem mass spectrometry.Anal.Chem.79, 3416–3424.https://doi.org/10.1021/ac0618674 (2007).
- Deng, P., Chen, X., Zhang, G. & Zhong, D. Bioanalysis of an oligonucleotide and its metabolites by liquid chromatography–tandem mass spectrometry.J. Pharm.Biomed.Anal.52, 571–579.https://doi.org/10.1016/j.jpba.2010.01.040 (2010).
- Gau et al.Oligonucleotide mapping via mass spectrometry to enable comprehensive primary structure characterization of an mRNA vaccine against SARS‑CoV‑2.Nature Scientific Reports.13:9038 2023.
- Loverix S, Winqvist A, Stromber R, Steyaert J. Mechanism of RNase T1: concerted triester-like phosphoryl transfer via a catalytic three-centered hydrogen bond Chem Biol.2000 Aug, 7(8):651-8 doi: 10.1016/s1074-5521(00)00005-3.
- Loverix S, Steyaert J. Deciphering the Mechanism of RNase T1.Methods in Enzymology, Academic Press, Vol.341, 2001, 305-323, doi: 10.1016/S0076-6879(01)41160-8
- Masachika Irie.3- RNase T1/RNase T2 Family RNases.Ribonucleases, Academic Press, 1997.101-130, doi: 10.1016/B978-012588945-2/50004-2.
- Sorrentino, Salvatore.The Eight Human “Canonical” Ribonucleases: Molecular Diversity, catalytic properties, and special biological actions of the enzyme proteins.FEBS Letters.Vol 584, Issue 11, 2010, 2194-2200, doi: 10.1016/j.febslet.2010.04.018
- Gaye M.M. et al.使用Premier BEH 300 Å C18寡核苷酸分析专用柱通过IP-RP-LC-MS方法表征CRISPR单向导RNA.沃特世应用纪要, 720007897ZH, 2023年10月.
720008130ZH,2023年11月