使用Premier BEH 300 Å C18寡核苷酸分析专用柱通过IP-RP-LC-MS方法表征CRISPR单向导RNA
摘要
CRISPR/Cas系统编辑速度快、成本低且对动物应用的需求少,因此成为研究和治疗领域执行基因组编辑的首选平台。该系统可以在基因组DNA的精确区域改变基因组,这些区域由独立的单向导RNA序列编程。结合后的CRISPR核糖核蛋白复合物随后将裂解、改变和/或修饰靶位点。编辑的特异性与上样RNA分子的设计和质量直接相关。
离子对反相液相色谱与质谱联用(IP-RP-LC-MS)等先进的分析方法有助于确保CRISPR RNA的鉴定、纯度、完整性和预期修饰。本研究提出,采用一种由高性能表面和寡核苷酸批次测试过的WidePore BEH C18吸附剂制成的色谱柱,作为可靠分离完整sgRNA和酶解sgRNA的起点。该色谱柱的分离度高,可通过测量完整质量数和基于MSE的酶解组分测序直接进行分子分析。
优势
- Premier BEH C18 300 Å寡核苷酸分析专用柱非常适合分析完整sgRNA及核酸酶酶解的sgRNA组分
- 色谱分离度高,可区分完整RNA的失败序列和降解物,实现可靠的完整性和纯度评估
- 酶解组分和修饰末端可实现出色的IP-RP-LC分离,从而完成详细的分子表征
- 使用计算机模拟酶解计算器、waters_connect™、UNIFI™和CONFIRM Sequence应用程序进行准确质量数匹配和sgRNA寡核苷酸映射注释
简介
CRISPR(成簇规律间隔短回文重复序列)是一种基因组编辑系统,最初在细菌中作为一种针对入侵噬菌体的适应性免疫应答被发现1。天然存在的CRISPR序列由tracrRNA(反式激活CRISPR RNA)和一段称为crRNA的RNA短片段组成2。这两个寡核苷酸连接成单向导RNA (sgRNA)。除RNA外,该复合物还含有Cas蛋白(例如Cas9),这是一种核酸内切酶,与sgRNA协同作用,在靶DNA中引入双链断裂,用于可编程的基因组编辑(图1)3。 CRISPR/Cas-9核糖核蛋白(RNP)系统的编辑分三个重要步骤进行:(i)基因组靶位点识别,(ii)双链切割,以及(iii)修复4。 编辑的准确性和有效性取决于sgRNA特定靶向待修饰基因位点的所需位置的能力。不正确的匹配会导致脱靶效应和效率损失5。CRISPR研究引起了极大关注,多种Cas蛋白的工程设计以及多项临床试验旨在利用离体基因编辑细胞疗法和体内基因编辑来治疗疾病6.7。
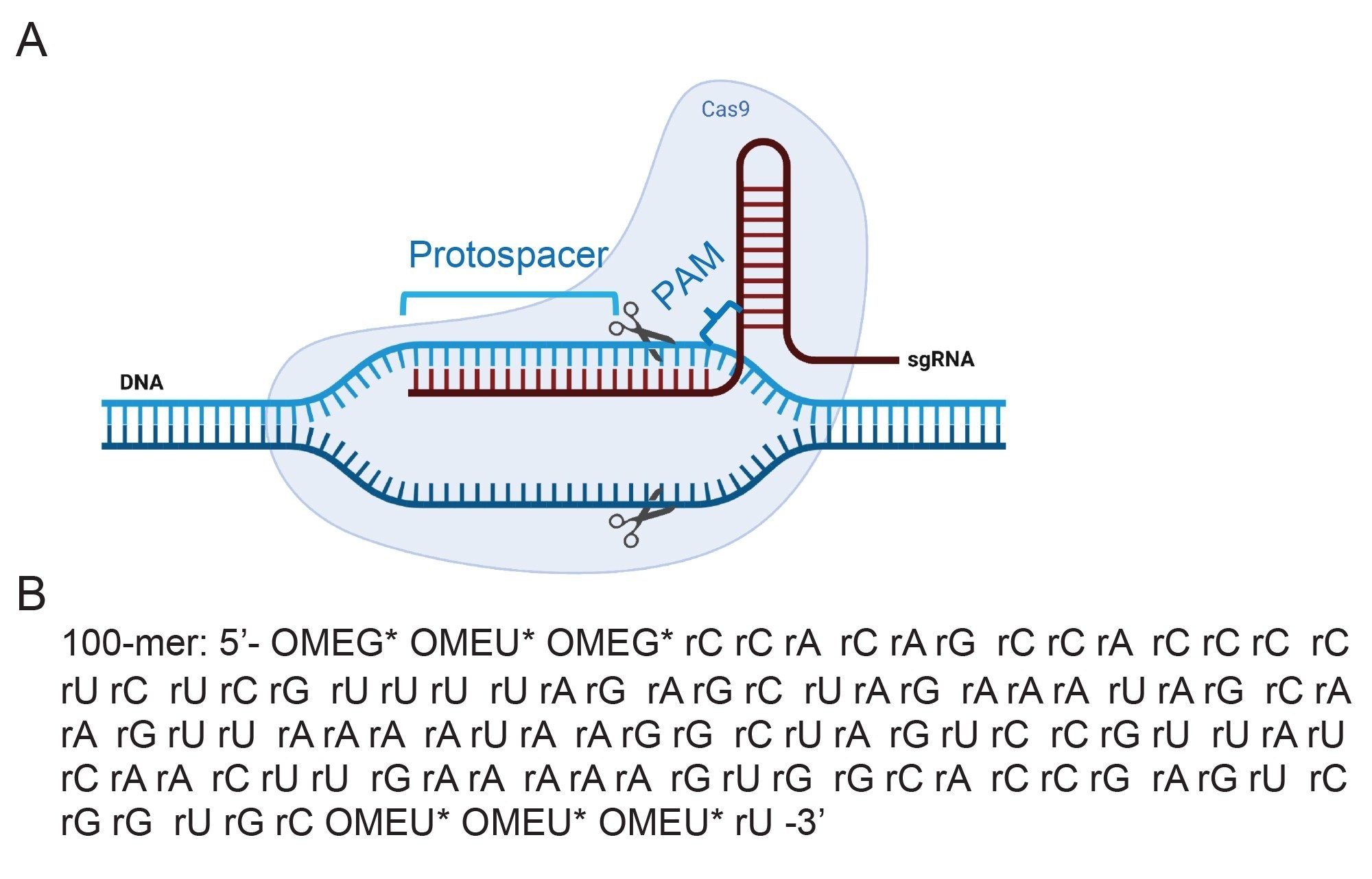 图1.(A) CRISPR-Cas9核糖核蛋白(RNP)复合物的示意图,其中包含与Cas9蛋白和互补DNA结合的单向导RNA (sgRNA)。图片来自BioRender.com。
图1.(A) CRISPR-Cas9核糖核蛋白(RNP)复合物的示意图,其中包含与Cas9蛋白和互补DNA结合的单向导RNA (sgRNA)。图片来自BioRender.com。(B)此分析中目标合成sgRNA的序列。OME=2'-甲基,*表示硫代磷酸酯连接子。sgRNA引导Cas-9通过5'crRNA识别靶DNA序列(原型间隔区),并在DNA解链并形成RNA-DNA杂合体后,在PAM序列上游区域(5'-NGG-3',N可以是任何核苷酸碱基)切割DNA。
由于可能存在脱靶效应,因此应仔细表征和监测sgRNA的序列、修饰和杂质。为此,可以通过液质联用法(LC-MS)进行完整RNA分析、寡核苷酸映射和测序。这些先进的方法可以直接观察RNA序列的分子结构,而基于互补DNA (cDNA)的Sanger测序和新一代测序(NGS)会丢失修饰信息,只能间接获取信息。本研究使用ACQUITY Premier BEH C18 300 Å, 1.7 µm色谱柱,在由LC、UV和MS仪器组成的单一平台上对完整和酶解RNA进行离子对反相液相色谱分析,使用基于应用程序的软件采集、处理数据并报告结果。该色谱柱采用的高性能表面技术和WidePore BEH颗粒可确保快速启动新的分离方法并实现完整sgRNA的可靠分离。当与寡核苷酸映射方法一起使用时,该色谱柱还有助于鉴定硫代磷酸异构体。本研究使用计算机模拟酶解库计算器(沃特世微应用商店)与waters_connect™(集成了UNIFI、CONFIRM Sequence和INTACT Mass应用程序)快速标注IP-RP-LC-MS数据。
实验
完整sgRNA分析
将小鼠GATA2 sgRNA(Integrated DNA Technologies,Coralville,Iowa;5’- OMEG* OMEU* OMEG* rC rC rA rC rA rG rC rC rA rC rC rC rC rU rC rU rC rG rU rU rU rU rA rG rA rG rC rU rA rG rA rA rA rU rA rG rC rA rA rG rU rU rA rA rA rA rU rA rA rG rG rC rU rA rG rU rC rC rG rU rU rA rU rC rA rA rC rU rU rG rA rA rA rA rA rG rU rG rG rC rA rC rC rG rA rG rU rC rG rG rU rG rC OMEU* OMEU* OMEU* rU -3’,OME=2'-O-甲基,*=硫代磷酸酯)重悬于不含核酸酶的Tris-EDTA缓冲液中,制得10 pmol/µL样品溶液。
液相色谱条件
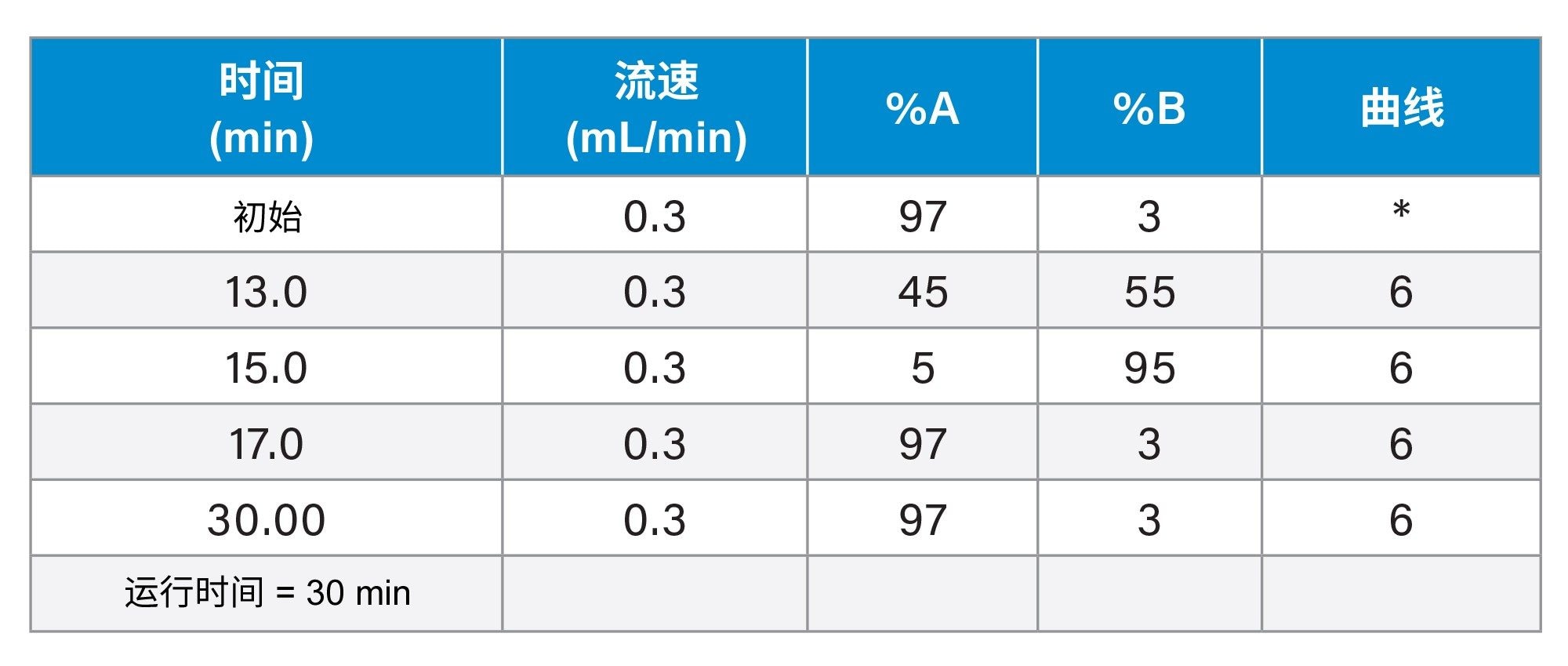
|
液相色谱系统: |
ACQUITY UPLC I-Class FTN(作为BioAccord™系统的一部分) |
|
检测器: |
ACQUITY UPLC TUV检测器 |
|
波长: |
260和280 nm |
|
色谱柱: |
ACQUITY Premier BEH C18, 300 Å, 1.7 µm, 2.1 × 100 mm寡核苷酸分析专用柱(p/n: 186010540) |
|
柱温: |
70 ˚C |
|
样品温度: |
4 ˚C |
|
进样: |
1 μL (10 pmol) |
|
流速: |
0.3 mL/min |
|
流动相A: |
含0.1% N,N-二异丙基乙胺(DIPEA)(作为IP试剂)和1% 1,1,1,3,3,3-六氟异丙醇(HFIP)的去离子水 |
|
流动相B: |
含0.0375% DIPEA和0.075% HFIP的65:35乙腈/水 |
梯度表
质谱条件
|
质谱系统: |
BioAccord LC-MS系统 |
|
检测器: |
ACQUITY RDa检测器 |
|
模式: |
包括碎片的全扫描 |
|
极性: |
负 |
|
锥孔电压: |
40 V |
|
碎裂锥孔电压: |
80-200 V |
|
质量范围: |
高(400–5000 m/z) |
|
扫描速率: |
2 Hz |
|
毛细管电压: |
0.80 kV |
|
脱溶剂气温度: |
400 °C |
寡核苷酸映射的样品前处理
使用24 µg(约10 kU)鸟苷酸特异性核糖核酸酶T1(Worthington Biochemical Corporation,新泽西州莱克伍德)酶解约90 µg小鼠GATA2 sgRNA (IDT)。首先,加入20 µL用不含核酸酶的缓冲液(10 mM Tris-HCl,pH 7.5,含0.1 mM EDTA)制备的8 M尿素,在80 °C下使RNA变性5 min。冷却至室温后,将核糖核酸酶T1重悬于不含核酸酶的缓冲液并加入变性RNA中,在37 °C下温育30 min。加入40 µL不含核酸酶的缓冲液,使酶解物达到80 µL,然后转移至聚丙烯自动进样器样品瓶(300 µL,p/n:186002639)中,使用分辨率为30 K的Q-Tof质谱仪(Waters Vion MS)进行IP-RP-LC-MS分析。
液相色谱条件
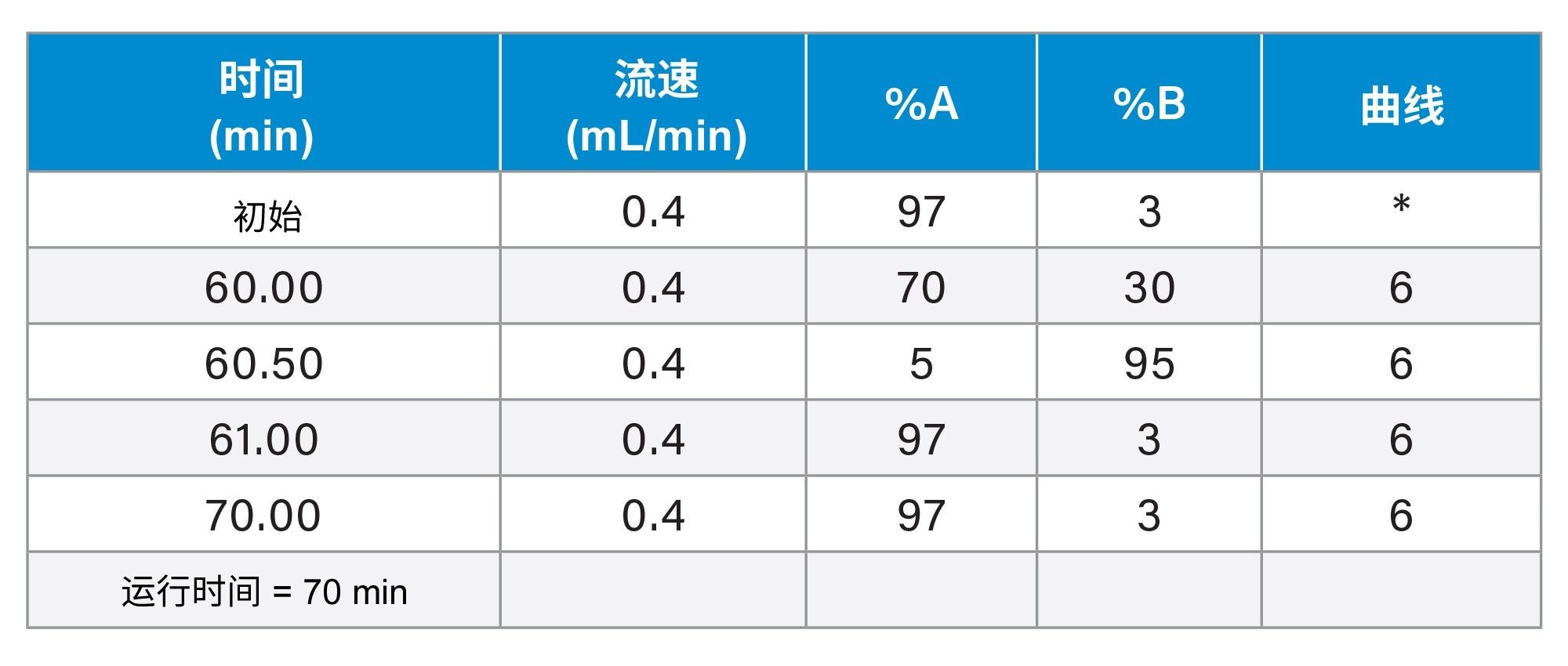
|
液相色谱系统: |
ACQUITY UPLC Premier BSM系统(作为BioAccord系统的一部分) |
|
检测器: |
ACQUITY UPLC TUV检测器 |
|
波长: |
260 nm |
|
色谱柱: |
ACQUITY Premier BEH C18, 300 Å, 1.7 µm, 2.1 × 150 mm寡核苷酸分析专用柱(p/n: 186010541) |
|
柱温: |
70 ˚C |
|
样品温度: |
4 ˚C |
|
进样: |
5 μL |
|
流速: |
0.4 mL/min |
|
流动相A: |
含0.1% N,N-二异丙基乙胺(DIPEA)(作为离子对试剂)和1% 1,1,1,3,3,3-六氟异丙醇(HFIP)的去离子水 |
|
流动相B: |
含0.0375% DIPEA和0.075% HFIP的65:35乙腈/水 |
梯度表
质谱条件
|
质谱系统: |
Vion MS系统 |
|
模式: |
包括碎片的全扫描 |
|
极性: |
负 |
|
锥孔电压: |
80 V |
|
毛细管电压: |
1.5 kV |
|
离子源温度: |
150 °C |
|
锥孔气流速: |
50 L/h |
|
脱溶剂气流速: |
1200 L/h |
|
脱溶剂气温度: |
600 °C |
|
低碰撞能量: |
6 V |
|
质量范围: |
高(50-4000 m/z) |
|
扫描速率: |
2 Hz |
|
高碰撞能量梯度开始: |
10 V |
|
高碰撞能量梯度结束: |
30 V |
|
碎裂锥孔电压: |
80-200 V |
|
启用智能数据捕获: |
是 |
信息学软件
mRNA酶解计算器可通过沃特世微应用商店获取,Intact Mass和Confirm Sequence应用程序集成于waters_connect中。
结果与讨论
将完整sgRNA进样至ACQUITY Premier BEH C18寡核苷酸分析专用柱(2.1 x 150 mm, 300 Å, 1.7 µm),然后使用DIPEA/HFIP改性的流动相和一段相对较短的梯度进行IP-RP-LC分离。质谱仪设置为在本次运行中采集低能量质谱图进行完整RNA质量数分析。还使用配备RDa高分辨率质量分析器和检测器的BioAccord LC-MS系统在负离子模式下采集了三次数据。该色谱柱所采用的WidePore固定相针对长度不超过200 nt的核酸分析进行了优化,因此非常适用于长度通常约为100个核苷酸的sgRNA分子。如图2所示,使用300 Å BEH C18色谱柱得到的总离子流色谱图(TIC)(时间范围为2~5.5 min)分离出除主要RNA组分以外的各种其他样品组分。4.95分钟处基峰的质谱图显示为多电荷离子包络。waters_connect Intact Mass应用程序去卷积显示分子量为32,242 Da,非常接近供应商提供的预期质量数值32,240 Da。该色谱法还分离了多种组分,例如sgRNA的失败序列或较短版本(保留时间更短)以及来自完整sgRNA的潜在加合物/延长。因此,该分离似乎非常适合检查合成sgRNA样品中存在的杂质的纯度、完整性、分离度以及最终鉴定结果。
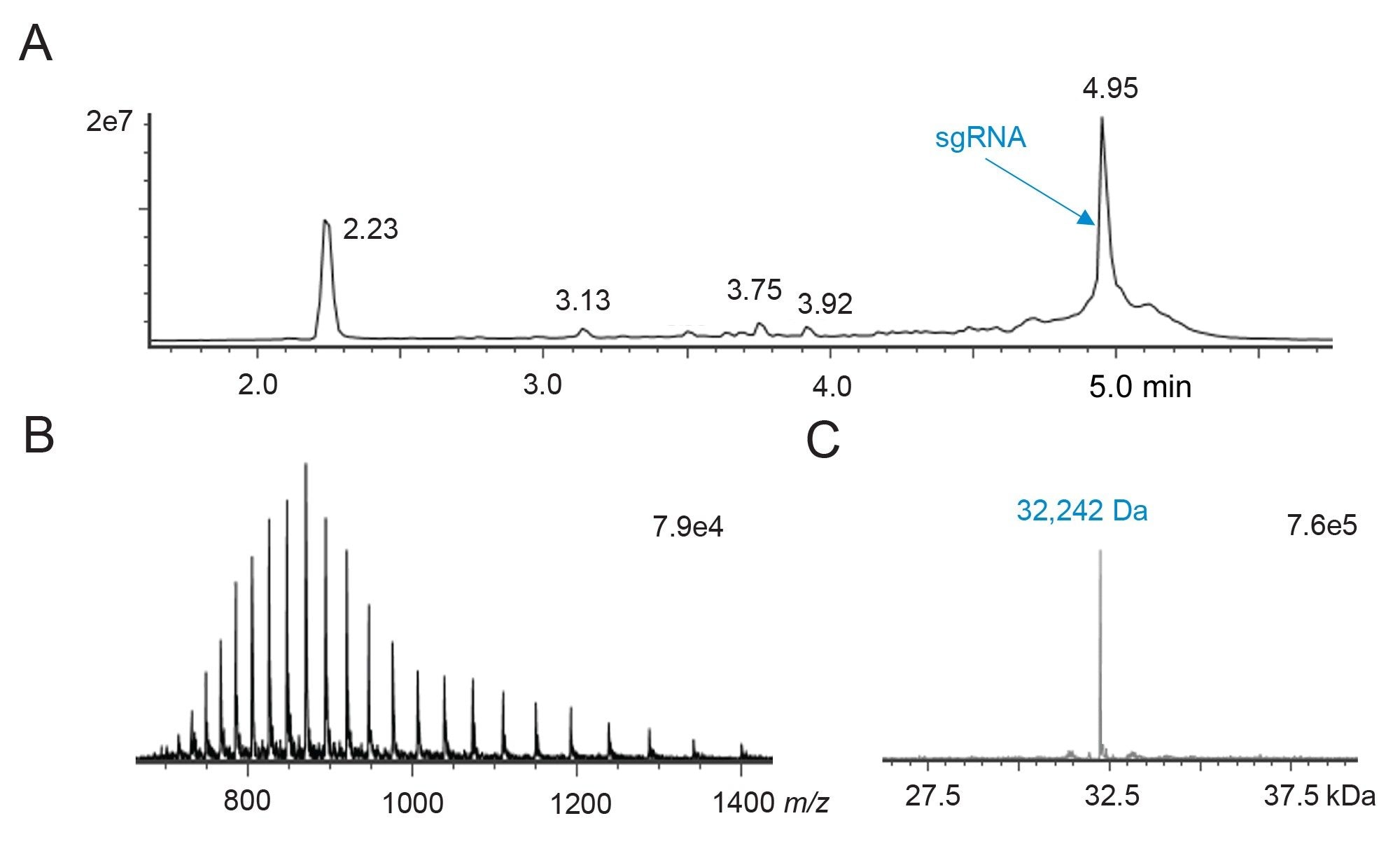 图2.10 pmol完整sgRNA的IP-RP-LC-MS分析结果。
图2.10 pmol完整sgRNA的IP-RP-LC-MS分析结果。(A) ACQUITY Premier BEH C18, 300 Å, 1.7 µm, 2.1 x 100 mm寡核苷酸分析专用柱和BioAccord系统采集的总离子流色谱图。
(B)保留时间4.95 min处峰的原始ESI负离子模式质谱图。
(C)完整sgRNA的去卷积质谱图和实测分子量32,242 Da(供应商报告的分子量为32240 Da)。
迄今为止,人们优先考虑通过合成方法生产sgRNA。此外,为提高抗降解能力,通常的做法是在5'-和3'-端添加带有2'-O-甲基(OME)的硫代磷酸酯(用*表示)核苷酸。我们考察了IP-RP-LC-MS鉴定和追踪这些修饰类型的能力。核糖核酸酶T1酶解和寡核苷酸映射的应用之前已有所描述[8]8。信息学方法从计算机模拟mRNA酶解计算器(沃特世微应用商店)开始,并使用该计算器来预测与核糖核酸酶T1酶解物对应的寡核苷酸酶解产物。指定了多个参数,包括“无修饰”、酶(核糖核酸酶T1)和允许的漏切(1个或0个)。计算器根据m/z范围、单同位素或平均质量数提供电荷态的默认值。将计算器生成的csv文件添加到UNIFI、waters_connect软件和CONFIRM Sequence应用程序中,以促进不同水平的峰鉴定。使用CONFIRM Sequence应用程序预测含硫代磷酸酯和2'-O-甲基基团的5'和3'端寡核苷酸,并将其手动添加到搜索空间中。总之,我们组建了一个包含各种分析物酶解产物的数据库,并将其导入UNIFI科学数据库中。除低能量离子扫描外,还将MS设定为采集高能量碎片离子谱图,以确证LC峰鉴定结果。本阶段的工作使用质量分辨率为30 K的QTof仪器。图3显示了用核糖核酸酶T1酶解的sgRNA的TIC谱图。在三次重复进样中,所得寡核苷酸酶解产物峰的保留时间差异极小(≤0.01 min)。所有预期的酶解产物均在3~47 min内洗脱。应用UNIFI中提供的HRMS筛选分析方法,通过筛选质谱图并在定义的容差限值内与分析物库匹配,来鉴定寡核苷酸酶解产物。手动验证质谱匹配,以检查质量精度、电荷态以及是否存在可提供序列信息的碎片离子。
仅包含磷酸二酯骨架的寡核苷酸酶解组分表现出尖锐且对称的峰,而含有硫代磷酸酯键的酶解组分观察到分裂峰,如图3B和图4所示。这些5'端和3'端物质的提取离子流色谱图证实,只有它们被分离为多峰模式,可能是由于非对映异构体分离。为确认每种寡聚物的序列,使用CONFIRM Sequence应用程序处理LC-MS数据,考虑至少500个强度计数,对MSE谱图中是否存在提供序列信息的子离子进行评分。图5展示了sgRNA的5'-端和3'-端酶解组分的鉴定结果,包括碎片离子的高可信度质量数匹配,可用于定位和计算硫代磷酸酯基团数量。这些匹配的点图表示进一步说明了观察到的碎片离子。
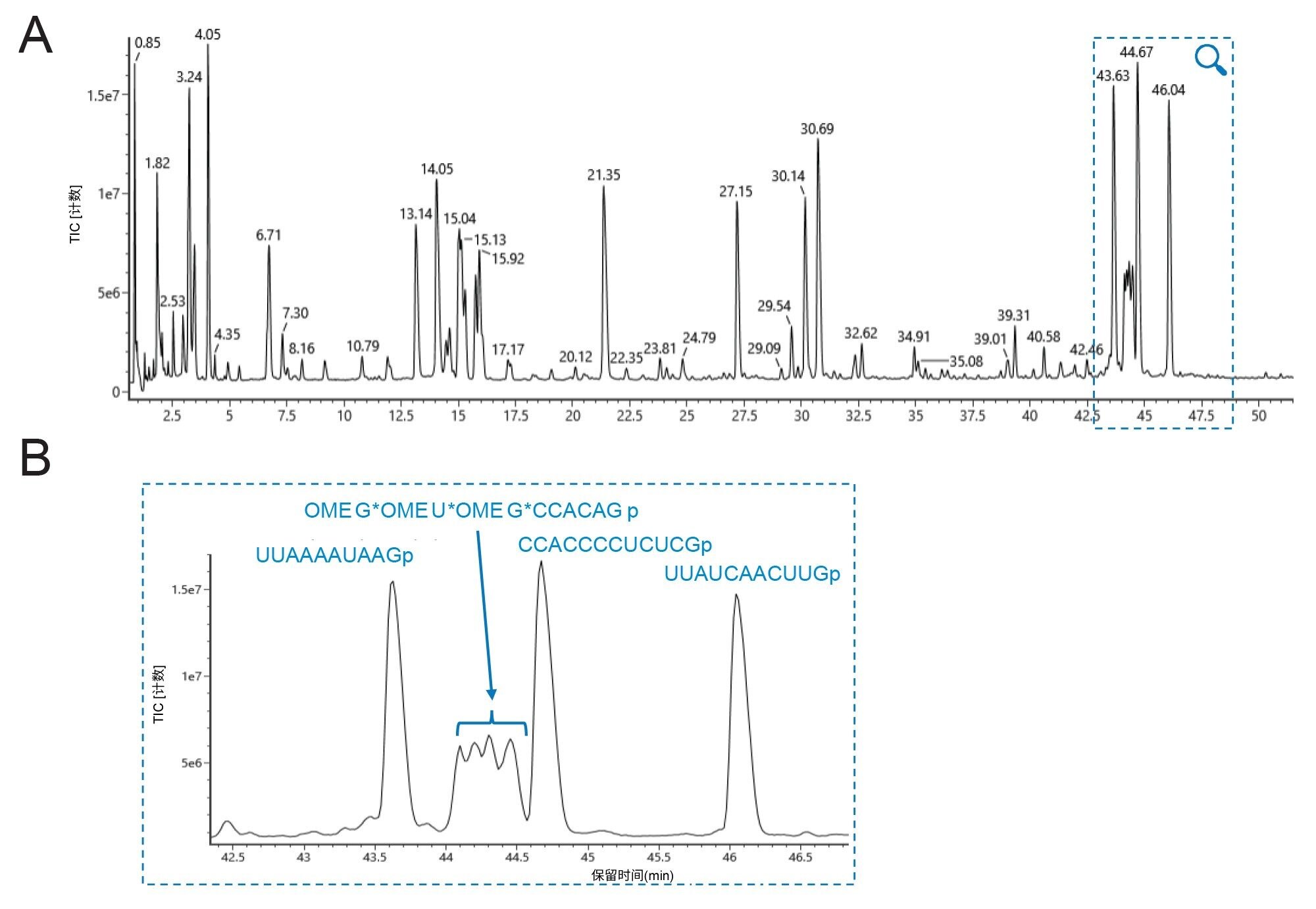 图3.使用ACQUITY Premier BEH C18, 300 Å, 1.7 µm, 2.1 x 100 mm寡核苷酸分析专用柱结合UV和QTof HRMS检测对由核糖核酸酶T1酶解的sgRNA进行IP-RP-LC-MS分析的寡核苷酸映射。(A) IP-RPLC总离子流色谱图(TIC)的全视图。
图3.使用ACQUITY Premier BEH C18, 300 Å, 1.7 µm, 2.1 x 100 mm寡核苷酸分析专用柱结合UV和QTof HRMS检测对由核糖核酸酶T1酶解的sgRNA进行IP-RP-LC-MS分析的寡核苷酸映射。(A) IP-RPLC总离子流色谱图(TIC)的全视图。(B) 33~47 min洗脱窗口的放大图和峰ID分配结果,包括含5'硫代磷酸酯的寡聚物OMEG* OMEU* OMEG* CCACAGp(位置1-9)(核糖的“r”注释已简化删除)。
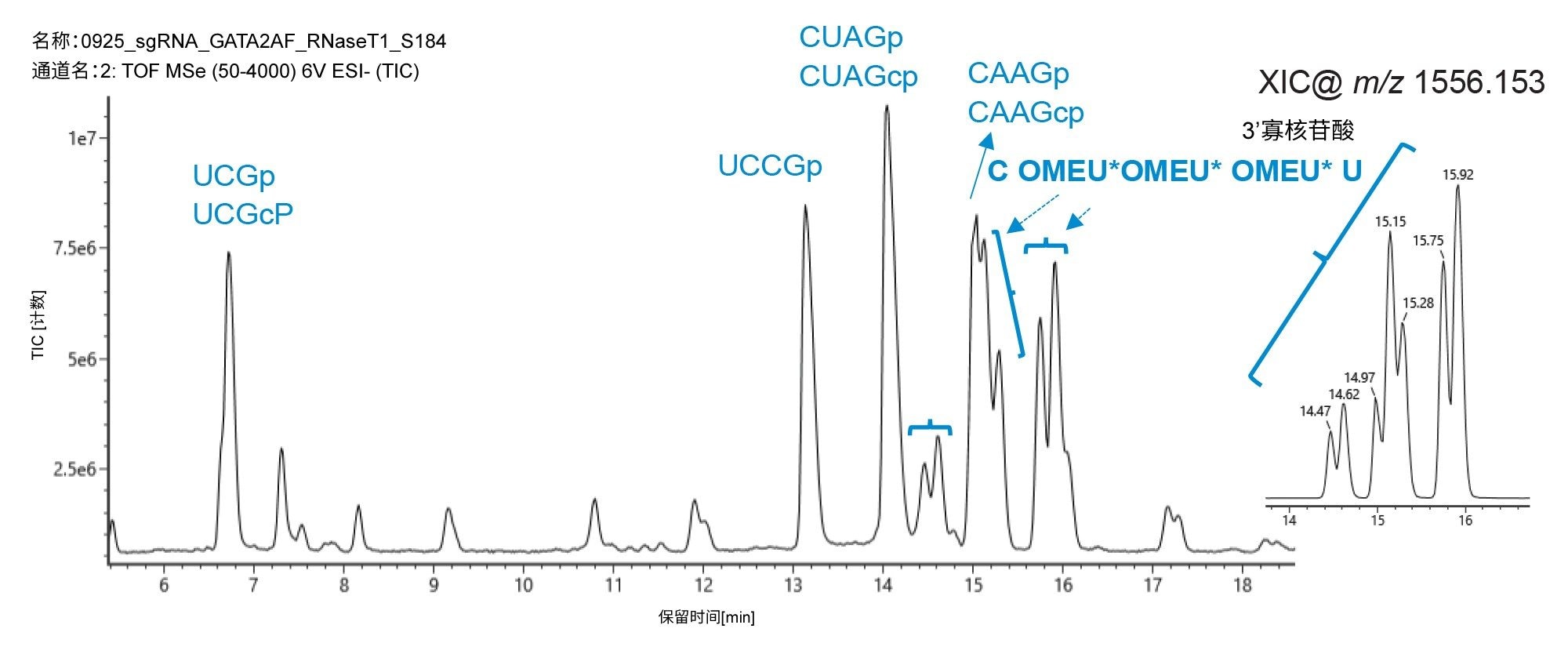 图4.使用ACQUITY Premier BEH C18, 300 Å, 1.7 µm, 2.1 x 100 mm寡核苷酸分析专用柱结合UV和QTof HRMS检测对由核糖核酸酶T1酶解的sgRNA进行IP-RP-LC-MS分析的寡核苷酸映射。6~18.5 min洗脱窗口的放大图和峰ID分配结果。RNA 3'端的非对映异构体与含硫代磷酸酯的寡聚体(C OMEU* OMEU* OMEU* U,位置96-100)用花括号表示。插图所示为m/z 1556.1537的提取离子流色谱图(XIC)。
图4.使用ACQUITY Premier BEH C18, 300 Å, 1.7 µm, 2.1 x 100 mm寡核苷酸分析专用柱结合UV和QTof HRMS检测对由核糖核酸酶T1酶解的sgRNA进行IP-RP-LC-MS分析的寡核苷酸映射。6~18.5 min洗脱窗口的放大图和峰ID分配结果。RNA 3'端的非对映异构体与含硫代磷酸酯的寡聚体(C OMEU* OMEU* OMEU* U,位置96-100)用花括号表示。插图所示为m/z 1556.1537的提取离子流色谱图(XIC)。
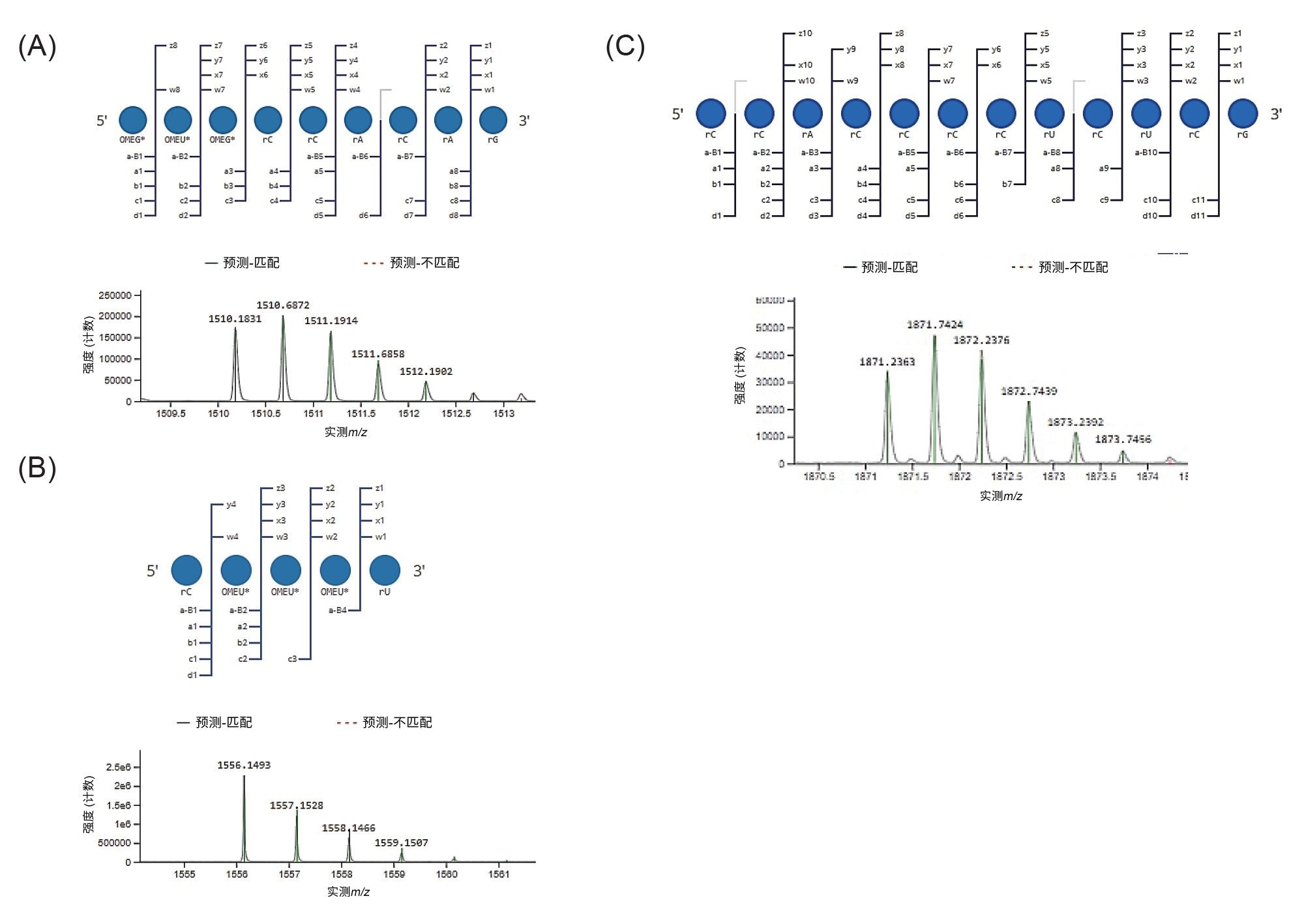 图5.5'端和3'端寡核苷酸的MS数据注释示例。图中展示了匹配的MSE碎片离子的点图以及采用同位素匹配后的低能量质谱图(以绿色突出显示)。5'-寡核苷酸酶解产物(位置1-9)OMEG* OMEU* OMEG* rCrCrArCrArG (A)、3'端寡核苷酸(位置96-100)rC OMEU* OMEU* OMEU* rU (B)和rCrCrArCrCrCrCrUrCrUrCrG对应于CONFIRM Sequence应用程序鉴定出的10–21 (C)之间的位置。
图5.5'端和3'端寡核苷酸的MS数据注释示例。图中展示了匹配的MSE碎片离子的点图以及采用同位素匹配后的低能量质谱图(以绿色突出显示)。5'-寡核苷酸酶解产物(位置1-9)OMEG* OMEU* OMEG* rCrCrArCrArG (A)、3'端寡核苷酸(位置96-100)rC OMEU* OMEU* OMEU* rU (B)和rCrCrArCrCrCrCrUrCrUrCrG对应于CONFIRM Sequence应用程序鉴定出的10–21 (C)之间的位置。
表1显示了在寡核苷酸母离子质量精度范围内(<7 ppm)检测到的酶解物组分列表,以及可为这些组分分配提供序列信息的碎片离子。至少需要80%的预期碎片离子(每个键至少有一个碎片离子)才能进行匹配。酶解物中显示出几种具有线性和环状磷酸盐形式的寡核苷酸。在sgRNA预期将得到的15种不同的理论酶解产物中,有三种组分(rUrGp、rArGp和rCrUrArGp)的序列来源可能不明确。其余的酶解组分各不相同,预计只会与序列中的一个位点匹配。因此,82%的序列可以通过独特的序列鉴定酶解组分进行鉴定。如果还考虑非独特的寡核苷酸,则覆盖率增加到98%。预计在完成核糖核酸酶T1酶解后释放的两个G残基代表此处sgRNA的最后2%。使用其他核酸酶和/或寻找T1漏切的实验方法可以实现100%的序列覆盖率。
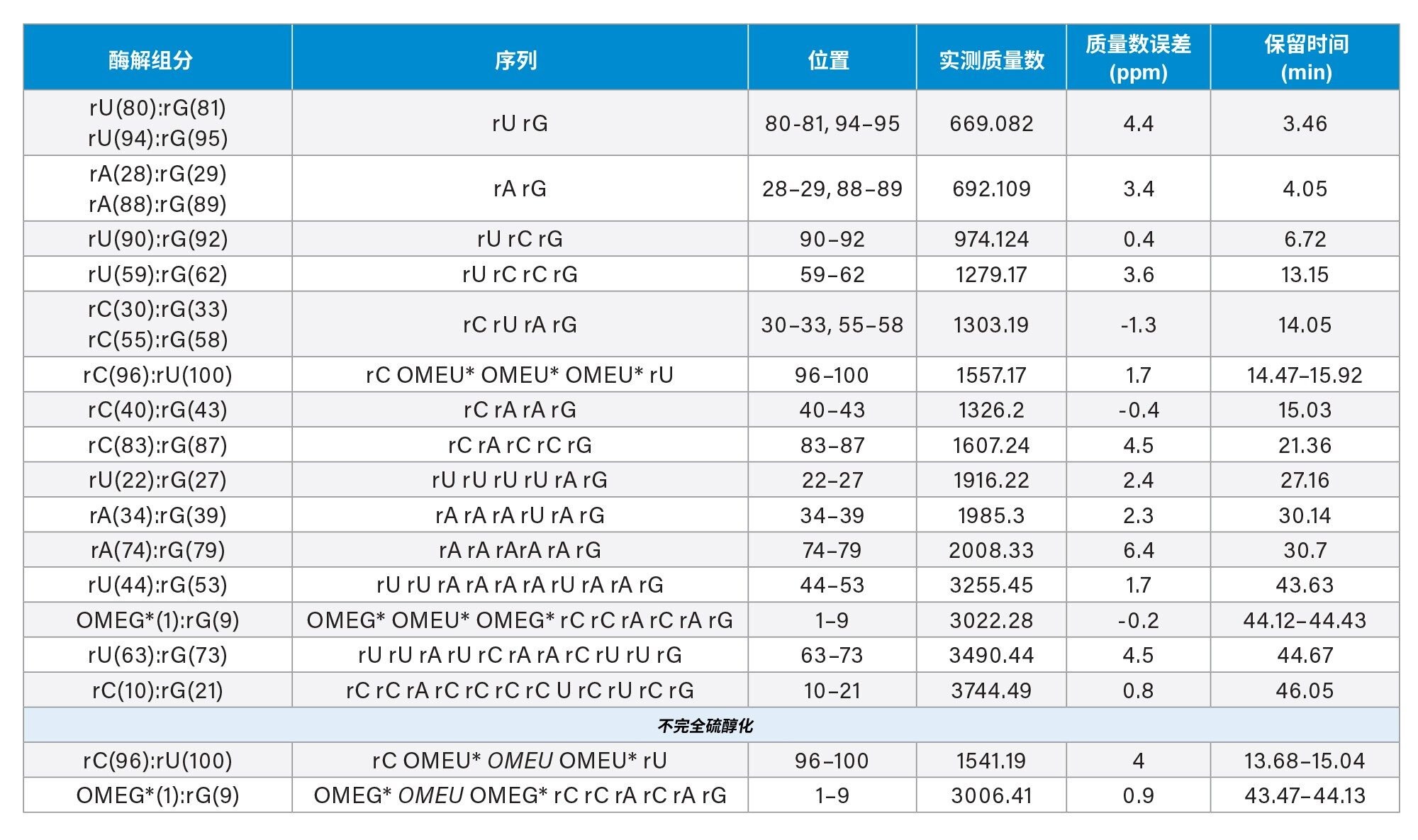 表1.使用UNIFI科学数据库搜索和waters_connect CONFIRM Sequence应用程序鉴定出的寡核苷酸酶解组分。通过将LC-MS谱图与预测的sgRNA核糖核酸酶T1酶解产物进行匹配,鉴定出第2列中列出的寡核苷酸。基于IP-RP-LC-MS检测sgRNA序列5'和3'端RNA骨架的不完全硫代磷酸化。*表示硫代磷酸酯连接子,#表示还鉴定出具有环磷酸酯封端的物质。在特定位置观察到的不完全硫醇化用核苷酸的斜体字表示。
表1.使用UNIFI科学数据库搜索和waters_connect CONFIRM Sequence应用程序鉴定出的寡核苷酸酶解组分。通过将LC-MS谱图与预测的sgRNA核糖核酸酶T1酶解产物进行匹配,鉴定出第2列中列出的寡核苷酸。基于IP-RP-LC-MS检测sgRNA序列5'和3'端RNA骨架的不完全硫代磷酸化。*表示硫代磷酸酯连接子,#表示还鉴定出具有环磷酸酯封端的物质。在特定位置观察到的不完全硫醇化用核苷酸的斜体字表示。
基于IP-RP-LC-MS检测sgRNA序列5’端和3’端RNA骨架的不完全硫代磷酸化。
IP-RP-LC-MS数据还显示,在sgRNA序列的5'和3'-区域均存在不完全硫醇化的寡核苷酸,如图6所示。在这些两次硫代磷酸化(具有预期的2'-O-甲基化)酶解组分中检测到大量信号(强度>2E4)。其母离子质量数的提取离子流色谱图产生了多峰模式,就像它们的3倍硫代磷酸化对应物一样。此外,MSE碎片离子数据中这些酶解组分的序列信息碎片离子的覆盖率>93%,表明它们的分配可信度高。这些寡核苷酸的保留时间不重叠,因此不是电喷雾诱导氧化的结果(表1)。非重叠谱图表明存在与合成相关的不完全硫醇化或与储存相关的硫-氧交换。
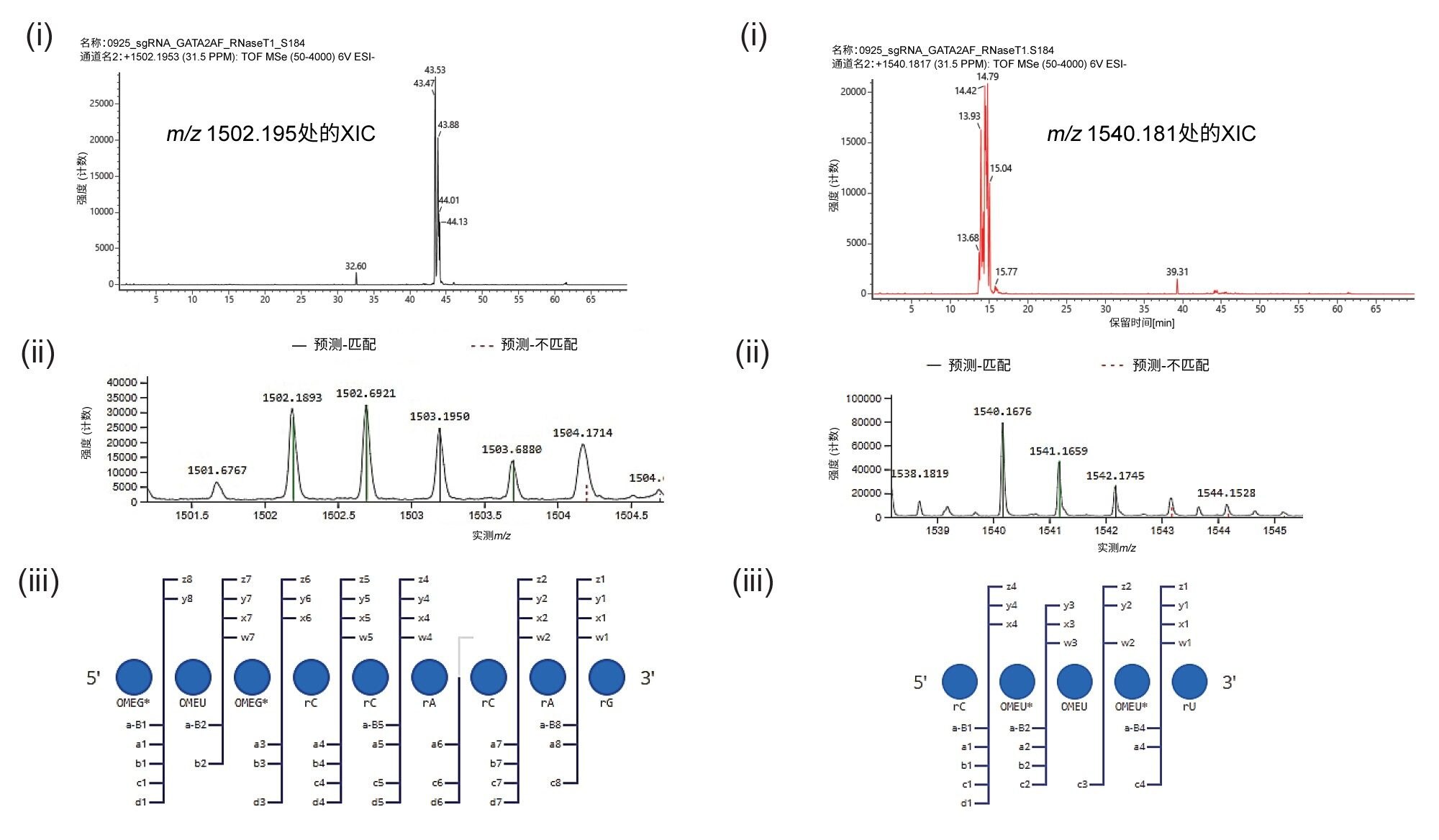 图6.基于IP-RP-LC-MS检测sgRNA序列5'和3'端RNA骨架的不完全硫代磷酸化。(A) 不完全硫醇化的5'端寡聚物OMEG* OMEU OMEG*rCrCrArCrArGp(位置1-9)的检测结果。(i) m/z 1502.1953的提取离子流色谱图(XIC)。(ii)通过CONFIRM Sequence匹配预期母离子寡核苷酸的同位素谱图。(iii)寡核苷酸碎片离子评分的点图,表明在尿苷处发生了不完全硫醇化。(B) 3'端寡核苷酸酶解产物rC OMEU* OMEU OMEU* rU(96-100位)的检测结果,具有不完全硫醇化。(i) m/z 1540.1817的XIC。(ii)通过CONFIRM Sequence匹配预期母离子寡核苷酸的同位素谱图。(iii)寡核苷酸的匹配MSE碎片离子评分点图,表明在尿苷处发生了不完全硫醇化。
图6.基于IP-RP-LC-MS检测sgRNA序列5'和3'端RNA骨架的不完全硫代磷酸化。(A) 不完全硫醇化的5'端寡聚物OMEG* OMEU OMEG*rCrCrArCrArGp(位置1-9)的检测结果。(i) m/z 1502.1953的提取离子流色谱图(XIC)。(ii)通过CONFIRM Sequence匹配预期母离子寡核苷酸的同位素谱图。(iii)寡核苷酸碎片离子评分的点图,表明在尿苷处发生了不完全硫醇化。(B) 3'端寡核苷酸酶解产物rC OMEU* OMEU OMEU* rU(96-100位)的检测结果,具有不完全硫醇化。(i) m/z 1540.1817的XIC。(ii)通过CONFIRM Sequence匹配预期母离子寡核苷酸的同位素谱图。(iii)寡核苷酸的匹配MSE碎片离子评分点图,表明在尿苷处发生了不完全硫醇化。
结论
本应用纪要介绍了一个全面表征sgRNA的示例,该示例可应用于基于CRISPR/Cas的基因组编辑试剂,这些试剂目前正准备用于医学、诊断和农业工程9。 本文展示了一种稳定的分析工作流程,可用于分析完整水平和酶解水平的sgRNA。用于IP-RP-LC-MS分析的ACQUITY Premier BEH C18 WidePore 300 Å色谱柱为这两种类型的分析提供了可靠的起点。
- 我们成功分析了合成sgRNA的完整性。
- 使用waters_connect Intact Mass应用程序通过去卷积得到sgRNA完整分子量。
- 无需样品净化即可快速获得sgRNA的核糖核酸酶T1寡核苷酸图谱。
- 该方法实现了高色谱分离度,因此可以通过MSE碎裂轻松分离和高效测序酶解组分。
- 通过精确质量数匹配(借助计算机模拟mRNA酶解计算器和waters_connectTM/UNIFI科学数据库应用程序),可以轻松注释生成的T1寡核苷酸图谱。
- 根据MSE谱图和waters_connect™平台的CONFIRM Sequence应用程序功能进一步验证分配的酶解组分序列,该功能提供了碎片离子覆盖率的直观显示,使结果易于解释。
- 根据诊断性非对映异构体分离,可以在数据中快速发现sgRNA的硫代磷酸化末端。此外,还可以区分和检测到这些物质的不完全硫醇化。
参考资料
- Barrangou, R., et al., CRISPR provides acquired resistance against viruses in prokaryotes.Science, 2007.315(5819): p. 1709-12.
- Deltcheva, E., et al., CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III.Nature, 2011.471(7340): p. 602-7.
- Jinek, M., et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity.Science, 2012.337(6096): p. 816-21.
- Shao, M., T.R. Xu, and C.S. Chen, The Big Bang of Genome editing technology: development and application of the CRISPR/Cas9 system in disease animal models.Dongwuxue Yanjiu, 2016.37(4): p. 191-204.
- Mohr, S.E., et al., CRISPR guide RNA design for research applications.Febs j, 2016.283(17): p. 3232-8.
- Pennisi, E., The CRISPR craze.Science, 2013.341(6148): p. 833-6.
- Charpentier, E., A. Elsholz, and A. Marchfelder, CRISPR-Cas: more than ten years and still full of mysteries.RNA Biology, 2019.16(4): p. 377-379.
- Maissa M. Gaye, J.F., Johannes P.C. Vissers, Ian Reah, Chris Knowles, and Matthew A. Lauber, 使用离子对液相色谱和质谱法进行合成mRNA寡核苷酸映射.沃特世应用纪要, 720007669ZH, 2022.
- Asmamaw, M. and B. Zawdie, Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing.Biologics, 2021.15: p. 353-361.
720007897ZH,2023年3月