Les personnes réalisant la caractérisation des polymères par des techniques chromatographiques n’utilisent pas exclusivement la GPC pour analyser leurs échantillons. Souvent, nous devons utiliser des techniques de chromatographie en phase liquide par adsorption ou la chromatographie de partage pour obtenir les informations requises.
Les techniques classiques de séparation en phase inverse et, parfois, en phase normale sont par exemple utilisées pour quantifier des additifs polymères. L’obtention de la distribution des masses moléculaires de l’échantillon polymère n’est parfois qu’une partie du processus de caractérisation. Qu’en est-il des additifs formulés dans le polymère pour stabiliser ou améliorer le traitement ? Ils peuvent être encore plus importants que le polymère lui-même. Nous devons penser à utiliser les bons stabilisants UV et les bons antioxydants pour éviter la dégradation, les bons plastifiants pour améliorer la souplesse, les bons antistatiques pour les polyoléfines, les bons retardateurs de flamme, les bons agents d’accélération pour améliorer le processus de réticulation (ou durcissement), etc.
Nous avons beaucoup travaillé sur les additifs polymères. Vous trouverez en détail certains de nos travaux dans le Journal of Liquid Chromatography, volume 14 n° 3 (1991) et volume 16, n° 7 (1993).
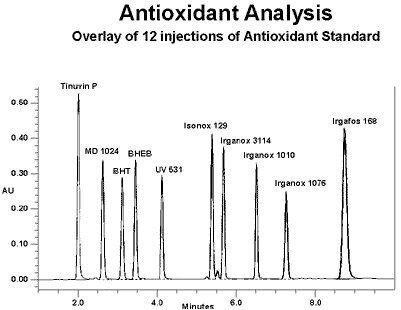
Comment analysons-nous les additifs polymères ? Tout d’abord, nous devons réfléchir à ce que nous tentons d’accomplir. Avons-nous besoin de savoir si les quantités correctes de chaque additif sont bien présentes dans la formulation ? Voulons-nous « déformuler » un matériau concurrent ? Devons-nous extraire le paquet d’additifs de la matrice polymère ? Il est probable que les réponses à ces questions soient « oui ». L’analyse par GPC n’est pas le meilleur moyen de séparer, d’identifier et de quantifier les niveaux d’additifs présents. La plupart des additifs sont assez proches les uns des autres en termes de taille et de masse moléculaire. Nous devons donc les séparer par HPLC. Une technique en gradient simple, avec programmation du débit en option, permet de séparer dans un délai d’analyse court de nombreux types d’additifs différents. Une analyse en gradient consiste à faire varier la composition de l’éluant ou de la phase mobile, généralement à partir d’un solvant « faible » jusqu’à un solvant « fort » sur une période donnée. Cette variation de composition s’effectue habituellement de façon linéaire pour l’analyse additive. Dans la mesure où nous faisons varier la composition de l’éluant tout au long de l’analyse chromatographique, le détecteur d’indice de réfraction ne peut pas être utilisé.
La plupart des additifs polymères que nous utilisons contiennent des chromophores qui absorbent la lumière ultraviolette. Un détecteur UV est donc principalement utilisé. En l’absence de chromophores, un détecteur évaporatif à diffusion de lumière peut être employé. Nous pouvons également modifier le débit tout au long de l’analyse, généralement en l’augmentant pour accélérer la sortie des éluants tardifs. La colonne habituellement choisie pour l’analyse additive est une colonne soit d’octadécylsilane (C18), soit d’octylsilane (C8) d’une longueur d’environ 15 cm. Voici un exemple de séparation par gradient en phase inverse (avec programmation du débit) d’une série d’antioxydants et d’agents stabilisants UV courants. Neuf (9) superpositions de 12 injections sont illustrées.



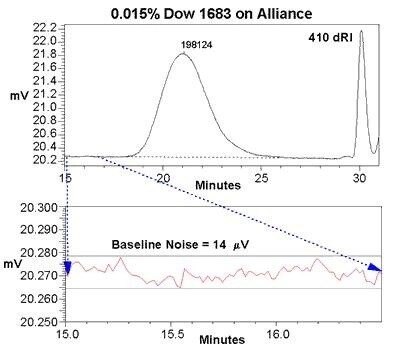
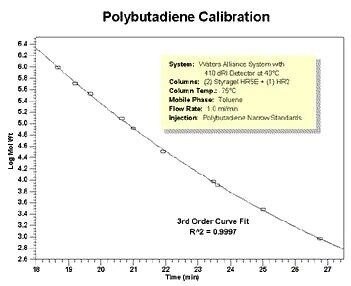




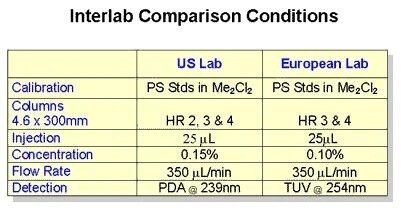








.jpg.82.resize/img.jpg)