CONFIRM Sequence: A waters_connect™ Application for Sequencing of Synthetic Oligonucleotides and Their Impurities
Abstract
This application note demonstrates an automated, compliance-ready liquid chromatography-mass spectrometry (LC-MS) workflow for sequence confirmation of oligonucleotides and their impurities.
Benefits
- CONFIRM Sequence is a novel waters_connect application developed for fast interpretation of complex tandem mass spectra (MS/MS) and MSE (no specific precursor selection) oligonucleotide mass spectra.
- The CONFIRM Sequence application is shown to be capable of providing complete sequence coverage (100%) for the full-length-product (FLP) as well as for low-abundance oligonucleotide impurities, as low as 0.2% abundance levels according to UV peak area measurements
- The CONFIRM Sequence app allows for fast data interpretation of complex oligonucleotide fragmentation spectra
- Data-independent acquisition (MSE or DIA) acquired for the FLP with an appropriate collision energy ramp provides complete sequence coverage without the need to perform collision energy optimization for individual oligonucleotide precursors
Introduction
Synthetic oligonucleotides are a class of therapeutics that emerged in the past decade as an alternative to small molecule and protein therapeutics.1–3 Manufacturing and quality control of oligonucleotide therapeutics requires highly selective and sensitive LC-MS methods. Ion pairing reversed-phase chromatography (IP-RP) is the most common LC-MS method used for characterizing synthetic oligonucleotides. Despite significant progress in solid phase oligomerization chemistry,4 synthetic oligonucleotides still contain multiple types of low-level (0.1–2%) impurities.3–5
An automated workflow for intact mass confirmation of synthetic oligonucleotides and their impurities employing the BioAccord™ System, operating under a compliance-ready environment, was previously described.6–9 In addition to intact mass confirmation, another cornerstone of complete oligonucleotide characterization is the sequence verification/validation process. Sequence accuracy is crucial for the activity of therapeutic oligonucleotides and other nucleic acid therapeutics (e.g. mRNA), as their nucleotide sequence is directly linked to their biological function within cells. Achieving a complete sequence coverage (100%) for unambiguous sequence assignment is desired. Following gas phase fragmentation of oligonucleotide precursors by tandem mass spectrometry, all the detected fragment ions are analyzed in order to confirm the expected oligonucleotide sequence. This can be a tedious, time-consuming process, when performed manually, because of the many possibilities of oligonucleotide fragmentation and the large variety of a, b, c, d, w, x, y, and z ions10 typically observed after collision-induced dissociation (CID) of oligonucleotide precursors. Due to this variety of fragment ions, combined with the presence of sequence non-informative fragments (such as the loss of nucleobases), the unambiguous sequencing of oligonucleotides is clearly more challenging than peptide sequencing.
Several computer programs have been developed over the last twenty years for automatic annotation of such complex gas phase dissociation spectra.11–18
CONFIRM Sequence is a recently introduced waters_connect application that automates the sequencing of synthetic oligonucleotides and their impurities, by automatically processing MS/MS spectra of oligonucleotides acquired via targeted MS/MS or untargeted MSE (DIA).
Here we investigated the capabilities of the CONFIRM Sequence application for fast, automatic sequencing of synthetic oligonucleotides and their impurities.
Experimental
Reagents and Sample Preparation
Triethylamine (TEA, 99.5% purity, catalogue number 65897-50ML) and methanol (LC-MS grade, catalogue number 34966-1L) were obtained from Honeywell (Charlotte, NC), while 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, 99% purity, catalogue number 105228-100G) was purchased from Sigma Aldrich (St Louis, MO). HPLC grade deionized (DI) type I water was purified using a MilliQ system (Millipore, Bedford, MA). Mobile phases were prepared fresh and used on the same day. A 21-mer heavily modified oligonucleotide, containing a 2’-OMe modification on 19 of its nucleosides, having the sequence GUA ACC AAG AGU AUU CCA UTT and the elemental composition C229H306N76O143P20 was purchased from ATDBio (Southhampton, UK). Stock solutions were prepared in DI water at a concentration of 1 µM (or 2.34 µg/mL), from which a 10 µL volume was injected, which corresponds to loading 10 picomoles of the 21-mer oligonucleotide on-column for MS/MS acquisitions.
LC Conditions
|
LC-MS System: |
Xevo™ G2-XS QTof coupled to H-Class Bio UPLC™ System |
|
Column: |
ACQUITY™ Premier OST Column 1.7 µm, 130 Å, 2.1 x 100 mm, (p/n: 186009485)
|
|
Column temp.: |
60 °C |
|
Flow rate: |
300 µL/min |
|
Mobile Phases |
|
|
Solvent A: |
40 mM HFIP (hexafluoroisopropanol), 7 mM TEA (triethylamine) in DI water, pH 8.6 |
|
Solvent B: |
20 mM HFIP, 3.5 mM TEA in 50% methanol |
|
Sample temp.: |
6 °C |
|
Sample vials: |
LC-MS certified, Maximum Recovery Vials (p/n: 186005663CV) |
|
Injection volume: |
10 µL |
|
Wash solvents |
|
|
Purge solvent: |
50% MeOH |
|
Sample Manager wash solvent: |
50% MeOH |
|
Seal wash: |
20% acetonitrile in DI water |
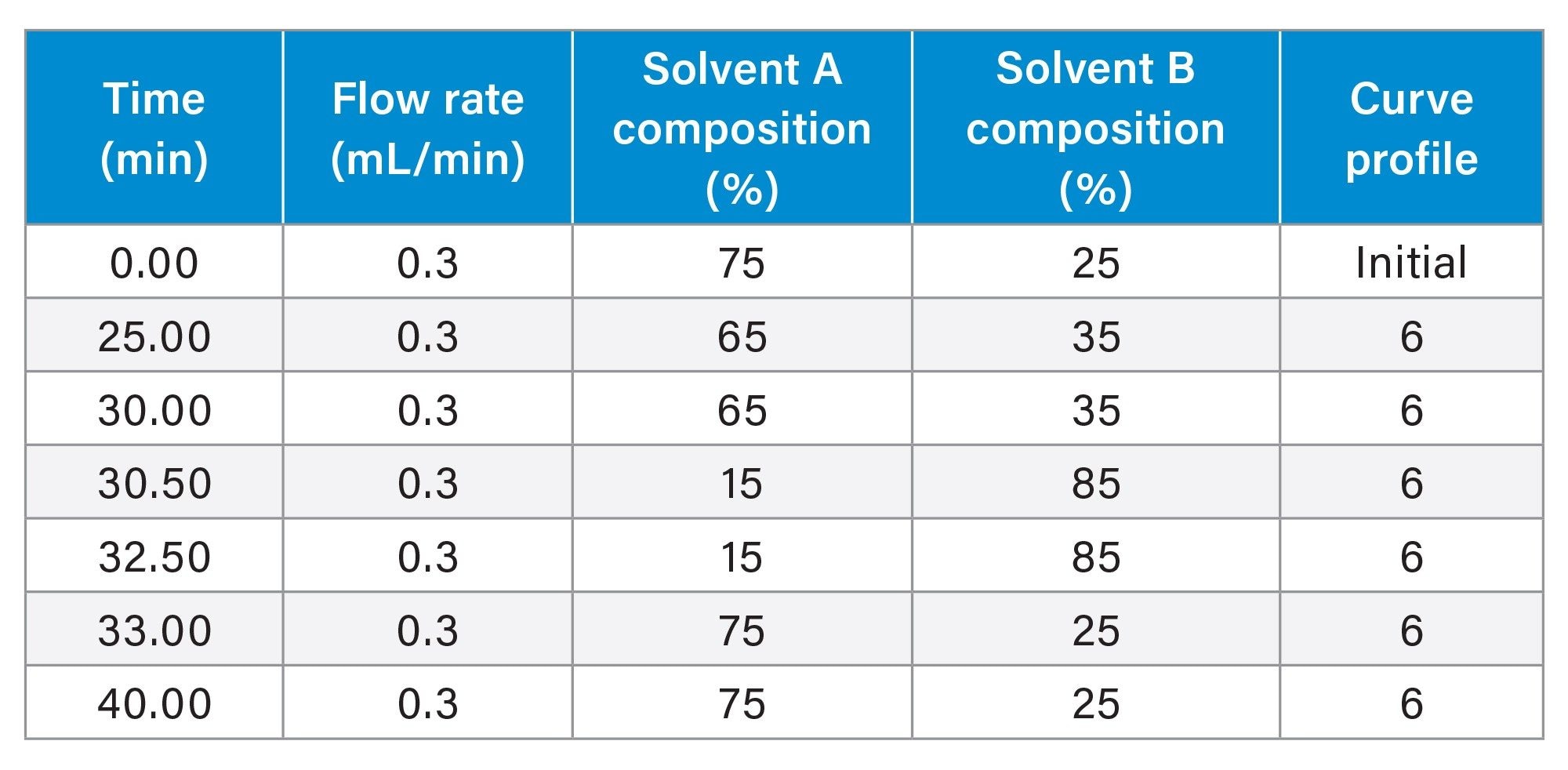
Gradient Table
MS Conditions
|
Capillary voltage: |
2.5 kV |
|
Cone voltage: |
40 V |
|
Source temp.: |
125 °C |
|
Desolvation temp.: |
400 °C |
|
Desolvation gas (N2) flow: |
600 L/h |
|
Cone gas flow: |
50 L/h |
|
TOF mass range: |
500–5000 |
|
Acquisition rate: |
1 Hz |
|
Collision energy: |
fifteen fixed collision energies in the range of 5–70 V (2, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, and 70 V) were investigated for targeted MS/MS acquisition of doubly, triply and quadruply charged oligonucleotide precursors; several high-energy CE ramps were investigated as well for MSE (DIA) acquisitions (20 to 40, 40 to 60, 60 to 80, and 80 to 100 V) |
|
Lock-mass: |
100 fmoles/µL GFP |
|
Data acquisition software: |
waters_connect 1.9.13.9 |
|
Data processing software: |
CONFIRM Sequence 1.0 |
Results and Discussion
A 21-mer oligonucleotide and its impurities was chosen as a test sample to demonstrate the capabilities and functionalities of the newly introduced CONFIRM Sequence application. A significant portion of the oligonucleotide contained modified nucleotides (19 out of 21 nucleotides) as illustrated in the sequence listed in the experimental section and in Figure 1. The 2’-OMe modification was attached to three guanosines (G) labeled in blue and seven adenosines (A) labeled in green in the oligonucleotide sequence. Besides the attachment of the same 2’-OMe functional group to uridines and cytidines, the nucleobases of these two nucleosides were further modified by the attachment of a 5-Methyl group to produce five 2’-OMe 5-Me uridines (U, labeled in purple) and four 2’-OMe 5-Me cytidines (C, labeled in red). The only nucleotides left unmodified are the two deoxythymidines (TT) at the 3’-end of the 21-mer.
An important component of the CONFIRM Sequence app is the Synthetic Library. The Synthetic Library allows the user to define the oligonucleotide sequence of interest. This library is divided into four individual libraries: Subcomponent Library, Monomer Library, Sequence Library and Modifier Library. Any individual nucleotide structure is broken down into three unique molecular entities: base, sugar, and linker, which are stored individually in the Subcomponent Library. These nucleotide building blocks are assembled into monomers that are kept separately in the Monomer Library. The user can combine a wide variety on nucleotide monomers into the desired oligonucleotide sequence which is saved in the Sequence Library. Sequences from the Sequence Library can be easily edited if desired. If the oligonucleotide contains non-standard (unusual) nucleotide modifications, these can be configured manually from the Modifier Library section. In the case of the 21-mer heavily modified oligonucleotide analyzed here, two monomers were configured in the Monomer Library because 2’-OMe-5-Me cytidine and 2’-OMe-5-Me uridine are not part of the default library. The screenshot displayed in Figure 1 shows how these two nucleotide monomers were assembled from their corresponding subcomponents. In the CONFIRM Sequence application, the sequence of the heavily modified 21-mer oligonucleotide is entered as: OMEG 2-OMe-5-MeU OMEA OMEA 2-OMe-5-MeC 2-OMe-5-MeC OMEA OMEA OMEG OMEA OMEG 2-OMe-5-MeU OMEA 2-OMe-5-MeU 2-OMe-5-MeU 2-OMe-5-MeC 2-OMe-5-MeC OMEA 2-OMe-5-MeU dT dT.
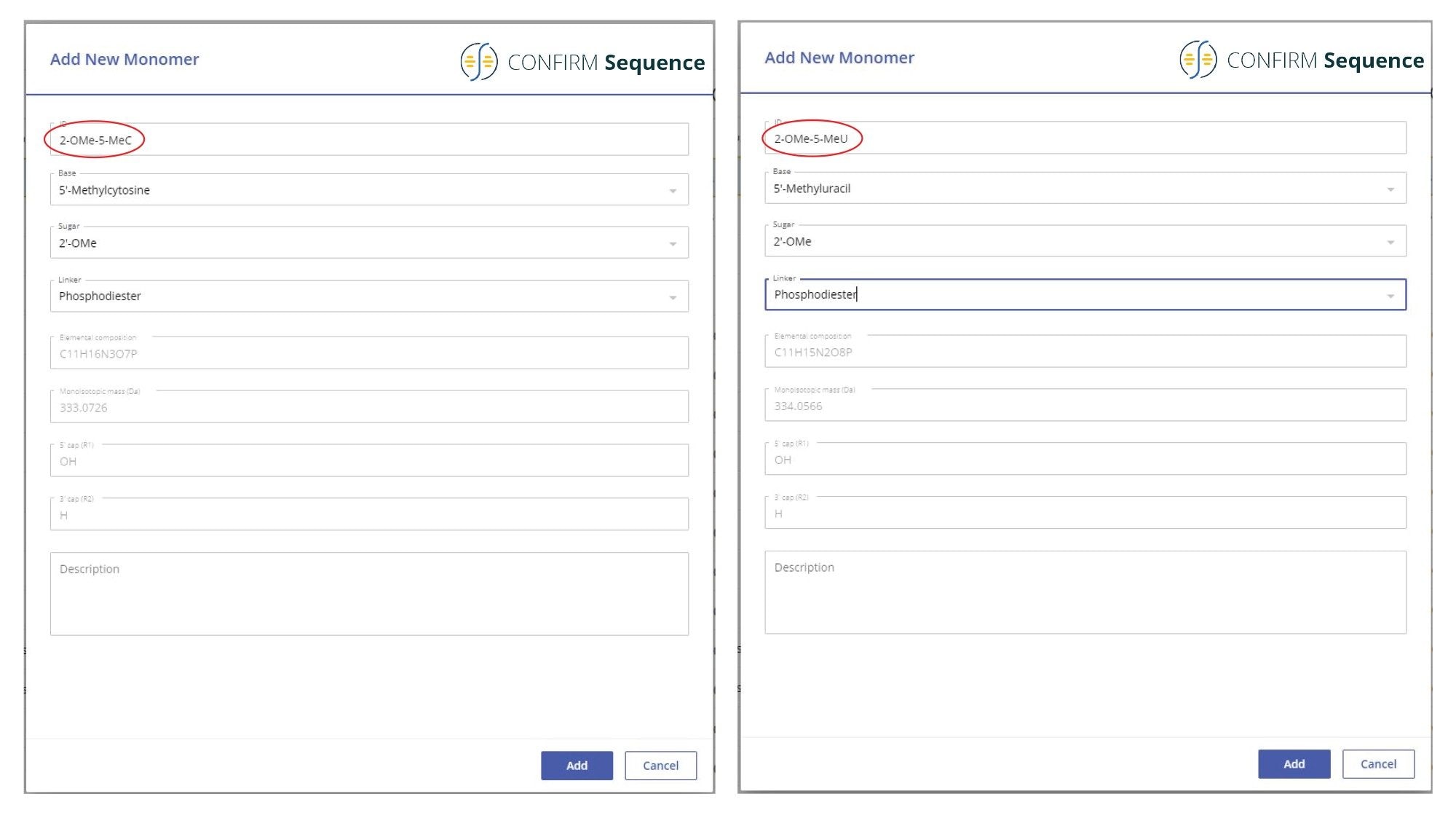 Figure 1. Monomer Library screenshots detailing how two nucleotide monomers (2’-OMe-5-Me cytidine and 2’-OMe-5-Me uridine) are created from their corresponding individual subcomponents (base, sugar and linker).
Figure 1. Monomer Library screenshots detailing how two nucleotide monomers (2’-OMe-5-Me cytidine and 2’-OMe-5-Me uridine) are created from their corresponding individual subcomponents (base, sugar and linker).
When analyzed on a 2.1 x 100 mm ACQUITY Premier OST Column (p/n: 186009485), the separation of the 21 nt oligonucleotide reveals a rather complex impurity profile as shown in Figure 2. Eleven oligonucleotide impurities were separated and detected by both TUV and MS detectors. For the first sample injection, 2 µL of the 1 µM 21-mer oligonucleotide were injected, and the data was acquired in negative ESI full scan mode (m/z=500–5000) to find the most abundant precursors corresponding to this oligonucleotide and its impurities (listed in Table I). In successive injections, 10 µL of sample were injected and the Xevo G2-XS instrument was operated in MS/MS mode using the quadrupole to isolate (with a ~5 Da window) certain multiply charged oligonucleotide precursors (see Table I). Following quadrupole isolation, each precursor underwent CID (collision-induced dissociation) fragmentation in the collision cell of the instrument to produce an MS/MS spectrum containing its corresponding fragment ions. A collision energy study was performed, with the applied voltages in the range of 10–80 V, to find the optimum fixed collision voltage that produced the best fragmentation (the higher sequence coverage of fragment ions) for each oligonucleotide. The CONFIRM Sequence app was used to evaluate the MS/MS sequence coverage for all MS/MS spectra recorded. In the case of the 21-mer oligonucleotide, a complete (100%) sequence coverage was obtained when its triply charged precursor ion ([M-3H]-3=2341.45) was fragmented with a fixed collision voltage of 63 V. The CONFIRM Sequence app uses a dot-map visual display, making it easy to assess the coverage of a predicted sequence, as illustrated in the screenshot presented in Figure 3. According to this figure, a wide variety of fragment ions were detected and assigned following the oligonucleotide fragmentation nomenclature.10 For data analysis, CONFIRM Sequence is using an in-house generated Targeted Isotope Clustering algorithm to match predicted oligonucleotide fragments with those detected in the raw data. The software displays the relevant matching information (graphically and in table format) and provides statistical analysis on each matched fragment. The dot-map display allows one to easily assess the coverage of a predicted sequence, or to locate an impurity modification. The CONFIRM Sequence app also allows the user to inspect the raw MS/MS fragmentation spectrum displayed in Figure 4. A great majority of the ion signals from this spectrum are colored in green in this figure, indicating that they have been matched to predicted fragment ions. The same maximum sequence coverage (100%) was obtained for the same oligonucleotide when a sample was analyzed using MSE acquisition. In this case, data-independent (without precursor isolation) was acquired using alternating one second MS scans starting with a low collision energy for precursor detection (10 V), followed by a collision energy ramp (40 to 60 V) to induce CID fragmentation of all precursors simultaneously. The dot-map results from the CONFIRM Sequence app displayed in Figure 5, demonstrates the capability of the MSE acquisition to produce high quality fragmentation data in a single sample injection, without the need for collision energy optimization. A relatively high sequence coverage (>70%) can be obtained using the same type of data acquisition (MSE) on a BioAccord Instrument (a small benchtop TOF MS system) as shown by the processing result from Figure 6. Compared to the data presented in Figures 3 and 5, the sequence coverage is lower for BioAccord acquisitions because several low m/z fragment ions are outside the acquired mass range of m/z=400–5000 Da.
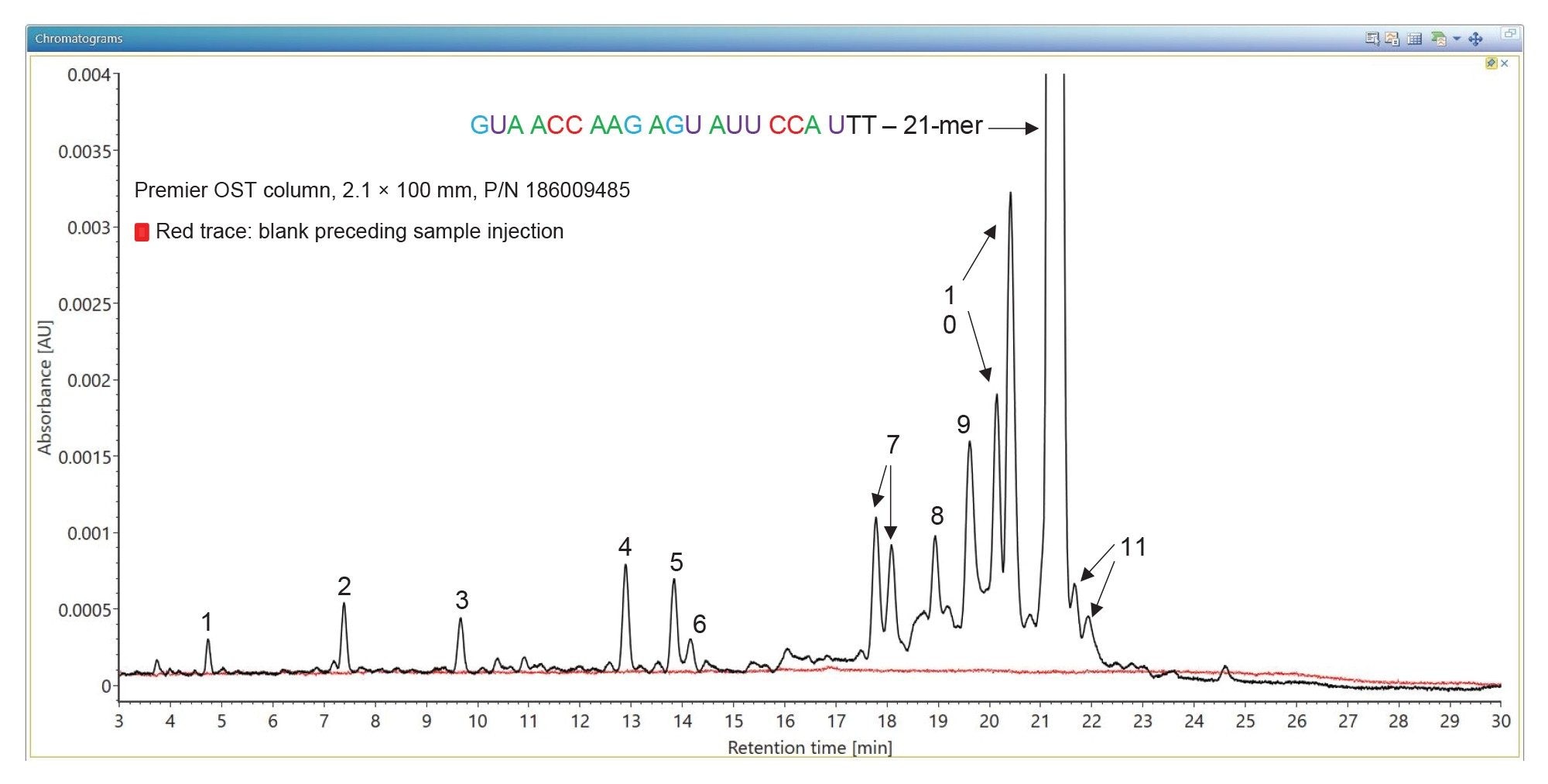 Figure 2. TUV chromatograms showing the separation of oligonucleotide impurities from a 21-mer heavily modified oligonucleotide. The Premier OST column resolves scrambled oligonucleotide sequences (like the doublets corresponding to peaks 7 and 10) and deamination isomers of the full-length product (peaks 11).
Figure 2. TUV chromatograms showing the separation of oligonucleotide impurities from a 21-mer heavily modified oligonucleotide. The Premier OST column resolves scrambled oligonucleotide sequences (like the doublets corresponding to peaks 7 and 10) and deamination isomers of the full-length product (peaks 11).
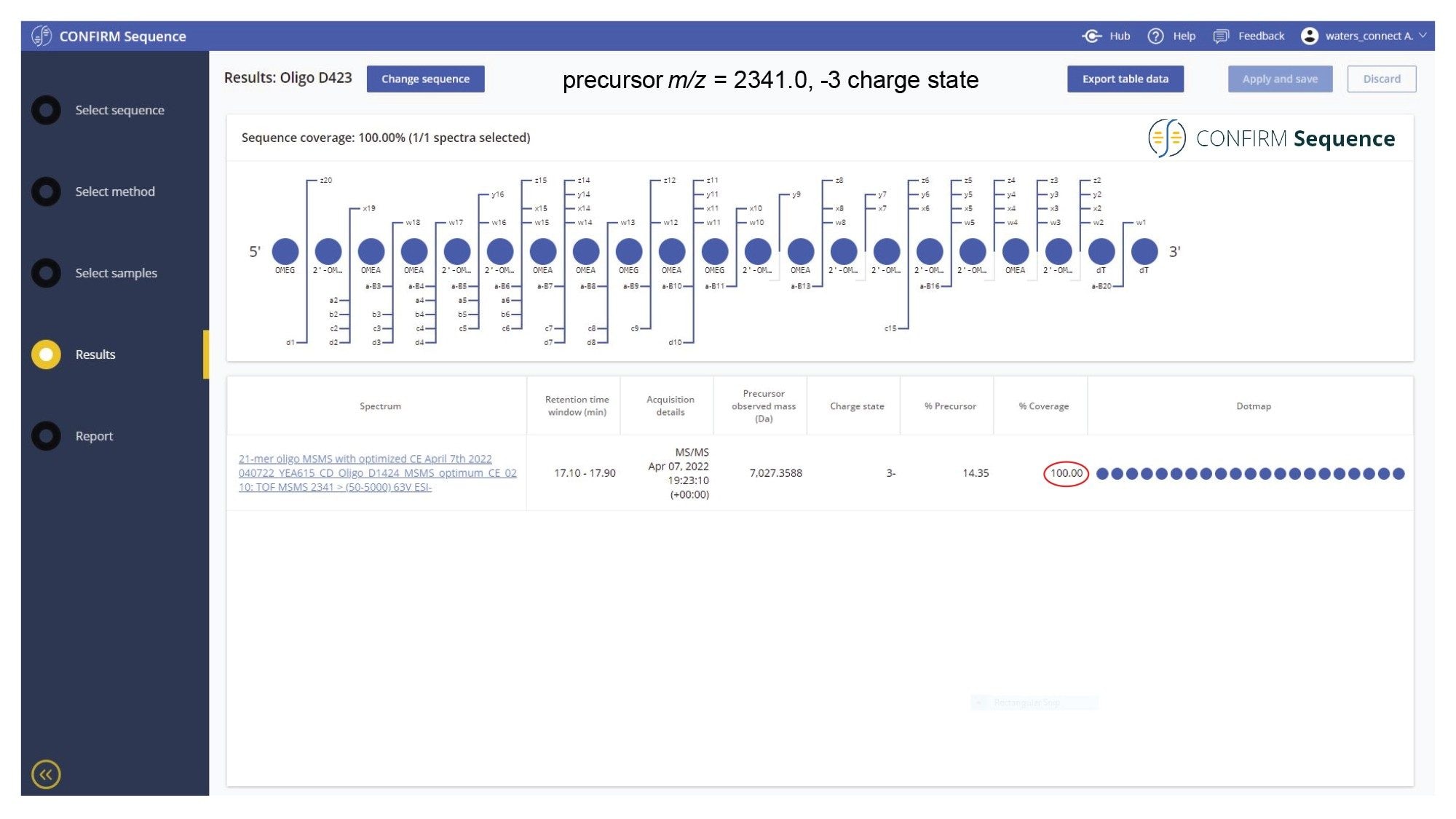 Figure 3. CONFIRM Sequence screenshot showing excellent MS/MS fragmentation coverage (100%) in a dot-map format. The [M-3H]-3 precursor of the 21-mer heavily modified oligonucleotide (m/z = 2342.0) was fragmented using an optimized fixed collision energy (set at 63 V) in the collision cell of a Xevo G2-XS QTof instrument.
Figure 3. CONFIRM Sequence screenshot showing excellent MS/MS fragmentation coverage (100%) in a dot-map format. The [M-3H]-3 precursor of the 21-mer heavily modified oligonucleotide (m/z = 2342.0) was fragmented using an optimized fixed collision energy (set at 63 V) in the collision cell of a Xevo G2-XS QTof instrument.
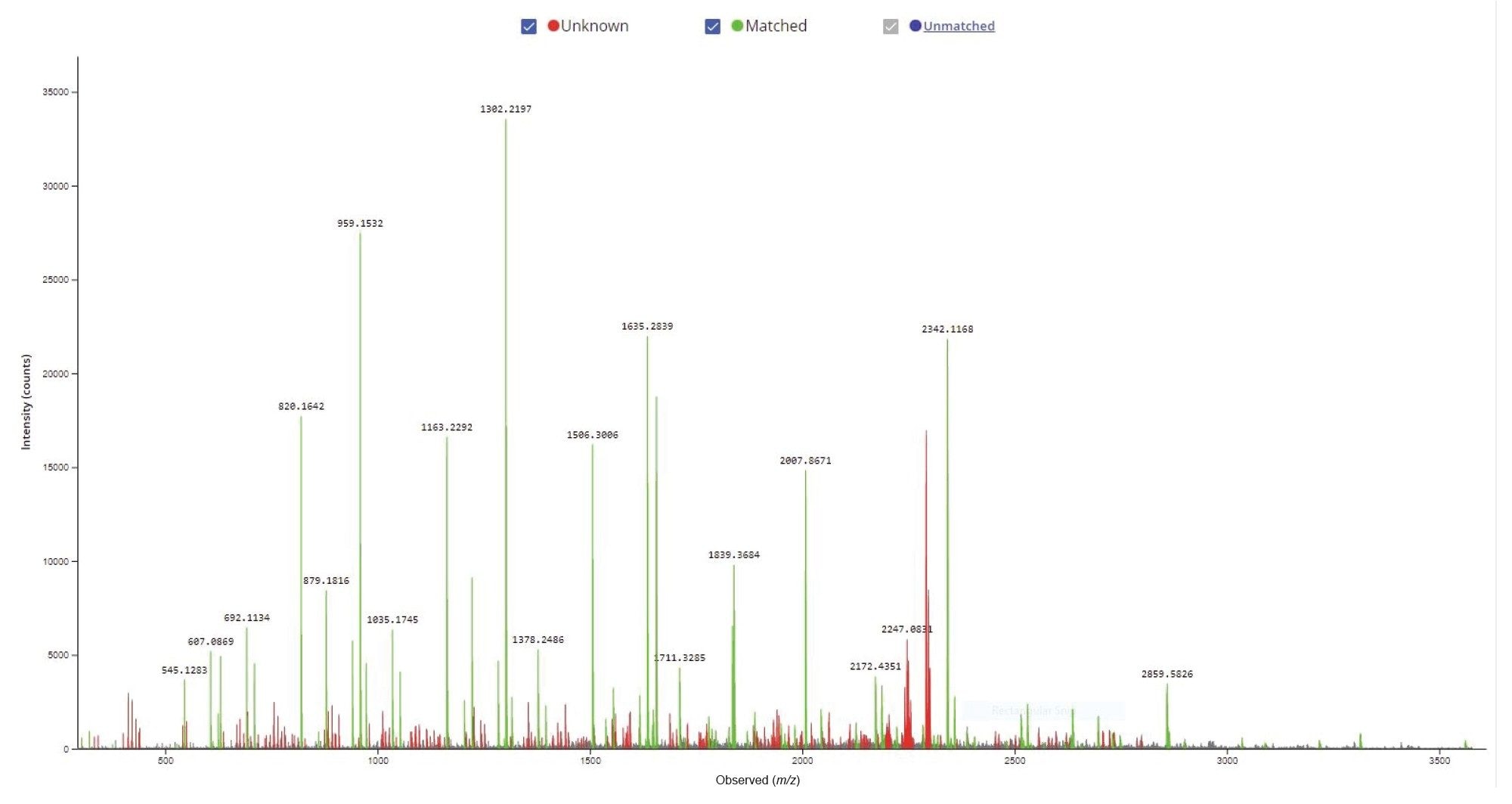 Figure 4. Confirm Sequence screenshot showing the MS/MS spectrum recorded for the 21-mer oligonucleotide. The [M-3H]-3 precursor of this oligo (m/z = 2341.0) was fragmented using an optimized fixed collision energy (set at 63 V) in the collision cell of a Xevo G2-XS QTof instrument. The fragment ions labeled in green were matched to the oligonucleotide sequence according to the dop-map diagram shown in Figure 3.
Figure 4. Confirm Sequence screenshot showing the MS/MS spectrum recorded for the 21-mer oligonucleotide. The [M-3H]-3 precursor of this oligo (m/z = 2341.0) was fragmented using an optimized fixed collision energy (set at 63 V) in the collision cell of a Xevo G2-XS QTof instrument. The fragment ions labeled in green were matched to the oligonucleotide sequence according to the dop-map diagram shown in Figure 3.
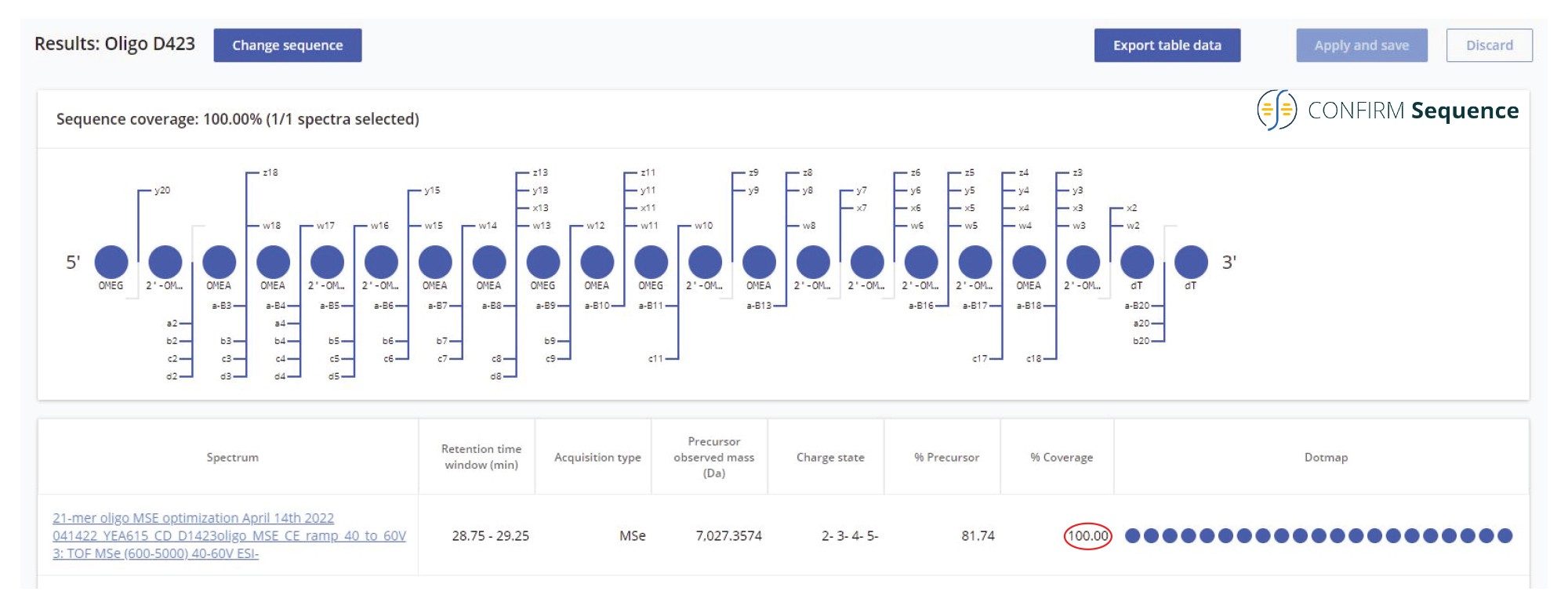 Figure 5. Complete (100%) sequence coverage, presented in a dot-map format, produced by high energy MSE untargeted fragmentation. All precursors of the 21-mer heavily modified oligonucleotide were fragmented using an optimized collision energy ramp (from 40 to 60 V) applied to the collision cell of a Xevo G2-XS instrument.
Figure 5. Complete (100%) sequence coverage, presented in a dot-map format, produced by high energy MSE untargeted fragmentation. All precursors of the 21-mer heavily modified oligonucleotide were fragmented using an optimized collision energy ramp (from 40 to 60 V) applied to the collision cell of a Xevo G2-XS instrument.
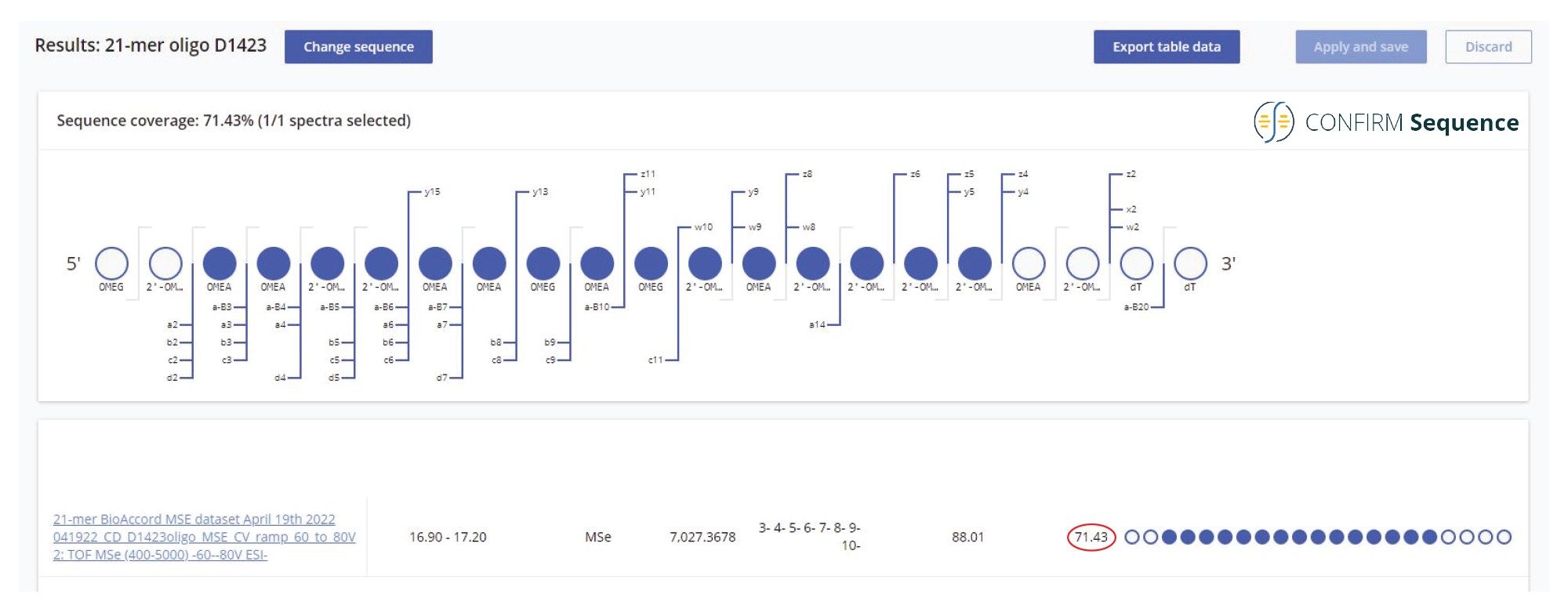 Figure 6. High sequence coverage (> 70%) obtained from high energy MSE untargeted fragmentation on BioAccord. All precursors of the 21-mer heavily modified oligonucleotide were fragmented using an optimized cone voltage ramp (from 60 to 80 V) applied to the Step Wave of a BioAccord Tof instrument.
Figure 6. High sequence coverage (> 70%) obtained from high energy MSE untargeted fragmentation on BioAccord. All precursors of the 21-mer heavily modified oligonucleotide were fragmented using an optimized cone voltage ramp (from 60 to 80 V) applied to the Step Wave of a BioAccord Tof instrument.
In addition to the full-length product (FLP), seven low-level impurities were sequenced on the Xevo G2-XS instrument using fixed collision energies, optimized for each oligonucleotide precursor according to the values presented in Table I.
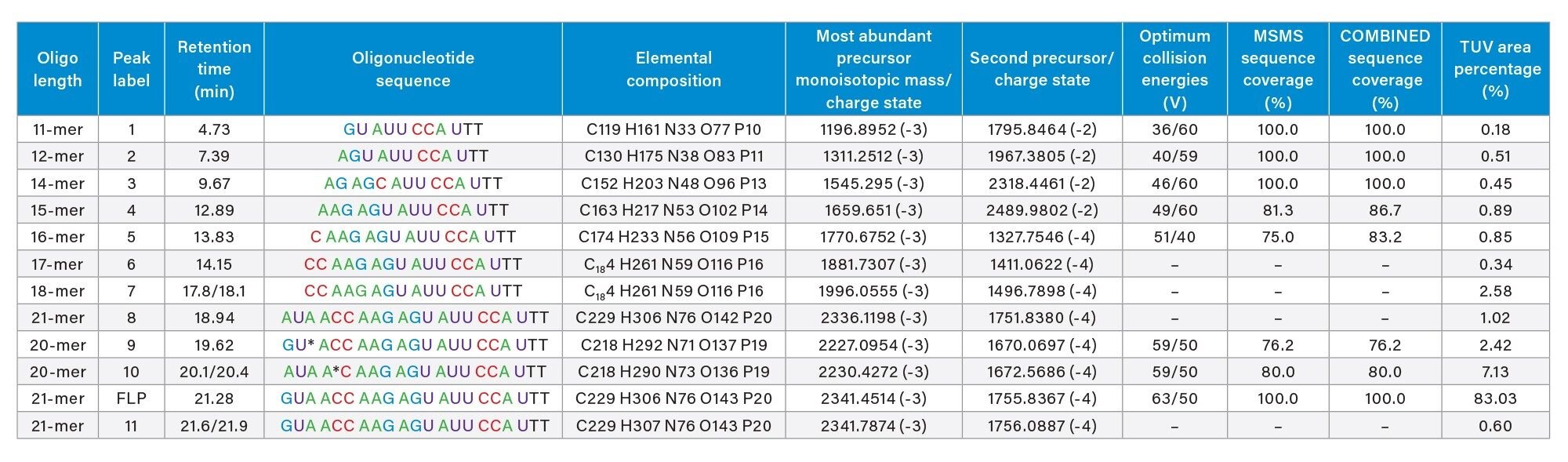 Table I. Eleven oligonucleotide impurities were identified in the 21-mer extensively modified oligonucleotide. Eight impurities and the full-length product (FLP) were sequenced using a Xevo G2-XS instrument and the individual MS/MS spectra fragmented with optimum collision energies were processed using the CONFIRM Sequence app. The MS/MS sequence coverage for the FLP and its impurities were above 75%. The lowest abundance impurity, an 11-mer oligonucleotide was fully sequenced (100% sequence coverage), while the sequence of the FLP (21-mer oligonucleotide) was also confirmed with 100% coverage. The total sequence coverage corresponds to the combined sequence obtained from the MS/MS fragmentation of two precursors of each oligonucleotide impurity.
Table I. Eleven oligonucleotide impurities were identified in the 21-mer extensively modified oligonucleotide. Eight impurities and the full-length product (FLP) were sequenced using a Xevo G2-XS instrument and the individual MS/MS spectra fragmented with optimum collision energies were processed using the CONFIRM Sequence app. The MS/MS sequence coverage for the FLP and its impurities were above 75%. The lowest abundance impurity, an 11-mer oligonucleotide was fully sequenced (100% sequence coverage), while the sequence of the FLP (21-mer oligonucleotide) was also confirmed with 100% coverage. The total sequence coverage corresponds to the combined sequence obtained from the MS/MS fragmentation of two precursors of each oligonucleotide impurity.
The lowest abundance impurity detected in this sample, corresponds to an 11-mer oligonucleotide present at 0.2% level based on UV peak area measurements. This 11-mer is eluting as the first chromatographic peak in the LC-UV chromatogram shown in Figure 2 and is labeled as peak 1 (see Figure 1 and Table I). The ESI-MS spectrum of this impurity indicates the presence of only two oligonucleotide precursors available for MS/MS fragmentation, as illustrated in Figure 7. Fragmentation of the most abundant precursor, the triply charged [M-3H]-3 ion at m/z=1196.89, using a fixed collision voltage of 39 V, produced a fragment-rich MS/MS spectrum. This great variety of fragment ions covered 100% of the sequence of this oligonucleotide impurity, as illustrated by the CONFIRM Sequence result displayed in Figure 8. The data presented in Figures 3 and 8 demonstrates that not only high abundant oligos (like the FLP 21-mer), but also low-abundance oligo impurities, as low as 0.2% abundance levels (the 11-mer impurity) when sequenced on the Xevo G2-XS QTof instrument can generate results with complete sequence coverage. The 11-mer oligo impurity, along with four other impurities (labeled as peaks 2–5) were all verified by the CONFIRM Sequence app to correspond to truncated versions of the 21-mer FLP missing a string of 5–10 nucleotides from the 5’-end of the parent oligo. These failed sequences are Class I impurities5 and are quite commonly present as by-products of the 3’-end to 5’-end oligonucleotide synthesis.3,4 The CONFIRM Sequence app is able to combine the results obtained from fragmentation of multiple precursors of the same oligonucleotide impurity in order to increase its sequence coverage. In this way, the sequence coverage for impurities labeled as peaks 4 and 5 was increased, as indicated by the COMBINED sequence coverage columns from Table I.
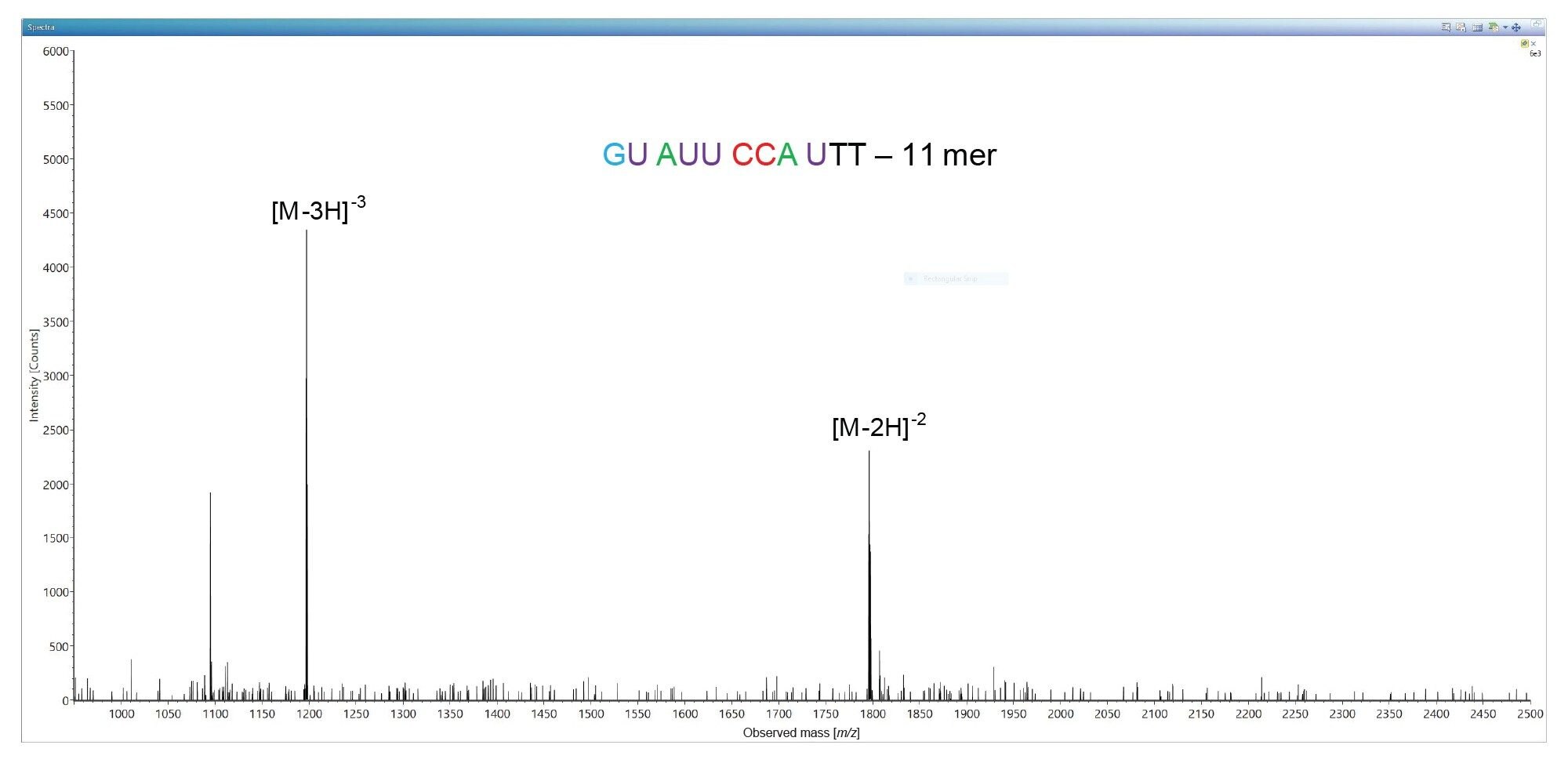 Figure 7. Ion pairing reversed phase ESI-MS spectrum of an 11-mer oligonucleotide impurity which is the least abundant impurity present in the 21-mer oligonucleotide sample (0.2% relative abundance, labeled as peak 1 in the chromatogram shown in Figure 1).
Figure 7. Ion pairing reversed phase ESI-MS spectrum of an 11-mer oligonucleotide impurity which is the least abundant impurity present in the 21-mer oligonucleotide sample (0.2% relative abundance, labeled as peak 1 in the chromatogram shown in Figure 1).
![Complete sequence coverage (100%) obtained from the MS/MS fragmentation of the [M-3H]-3 precursor of the 11-mer oligonucleotide impurity. The precursor was fragmented with an optimized fixed collision energy (36 V) in the collision cell of a Xevo G2-XS instrument](/content/dam/waters/en/app-notes/2022/720007677/720007677en-f8.jpg.82.resize/img.jpg) Figure 8. Complete sequence coverage (100%) obtained from the MS/MS fragmentation of the [M-3H]-3 precursor of the 11-mer oligonucleotide impurity. The precursor was fragmented with an optimized fixed collision energy (36 V) in the collision cell of a Xevo G2-XS instrument.
Figure 8. Complete sequence coverage (100%) obtained from the MS/MS fragmentation of the [M-3H]-3 precursor of the 11-mer oligonucleotide impurity. The precursor was fragmented with an optimized fixed collision energy (36 V) in the collision cell of a Xevo G2-XS instrument.
A different class of oligo impurities are the peak doublets identified as peaks 7, 10, and 11 in the chromatogram shown in Figure 2. In this case, each doublet corresponds to a pair of oligos that have identical (isobaric) precursors, therefore these impurities are expected to be sequence variants derived from the parent molecule and are Class III impurities.5 The last eluting doublet (peak 11) is very likely corresponding to the deaminated versions of the FLP and they are probably related to the presence of 2’-OMe-5-Me cytidines in the 21-mer sequence, which are prone to undergo deamination.19 It is remarkable that the ACQUITY Premier OST Column can resolve such small modifications (+1 Da) on the tail of the major oligo component as shown in Figure 2.
The most abundant impurities present in the 21-mer oligonucleotide are two sequence variants 20-mers, missing a 2’-OMe-5-Me cytidine, identified as the peak doublet 10 in Figure 2. There are 4 modified cytidines in the 21-mer sequence, but because they are located in pairs, there are only two possible 20-mer impurities missing a single 2’-OMe-5 Me cytidine. The CONFIRM Sequence app can search for sequence modifications (omissions or additions) in an attempt to match the MS/MS fragment ions of a known sequence and precursor mass. In this case, the software looked for a missed 2’-OMe-5 Me cytidine from the sequence of the 21-mer and found strong evidence (80% sequence coverage) for assigning this impurity to the sequence: GUA A*C AAG AGU AUU CCA UTT, with the missing nucleotide residue in position 5. This oligonucleotide sequence displayed a significantly higher sequence coverage (80%) compared with an alternative sequence which is missing the same residue at position 16 (which had only 50% sequence coverage). Both processing results are displayed in Figure 9. The MS/MS spectrum recorded for the most abundant sequence variant of the 20-mer pair (belonging to peak 10 doublet and eluting just before the FLP in Figure 1) is presented in Figure 10. The triply charged precursor of this oligo (m/z=2230.42) was fragmented using an optimized fixed collision energy (set at 59 V) in the collision cell of the Xevo G2-XS QTof Instrument. The a-B5 fragment ion detected at m/z=1506.29 confirms the presence of one 2’-OMe-5-Me cytidine close to the 5’-end of this oligo, while the w10 fragment ion present at m/z=1656.77 indicates that the other two 2’-OMe-5-Me cytidines are located at the other end of the molecule. In this way, the MS/MS spectrum from Figure 10 confirms the sequence of the most abundant impurity as: GUA AC AAG AGU AUU CCA UTT.
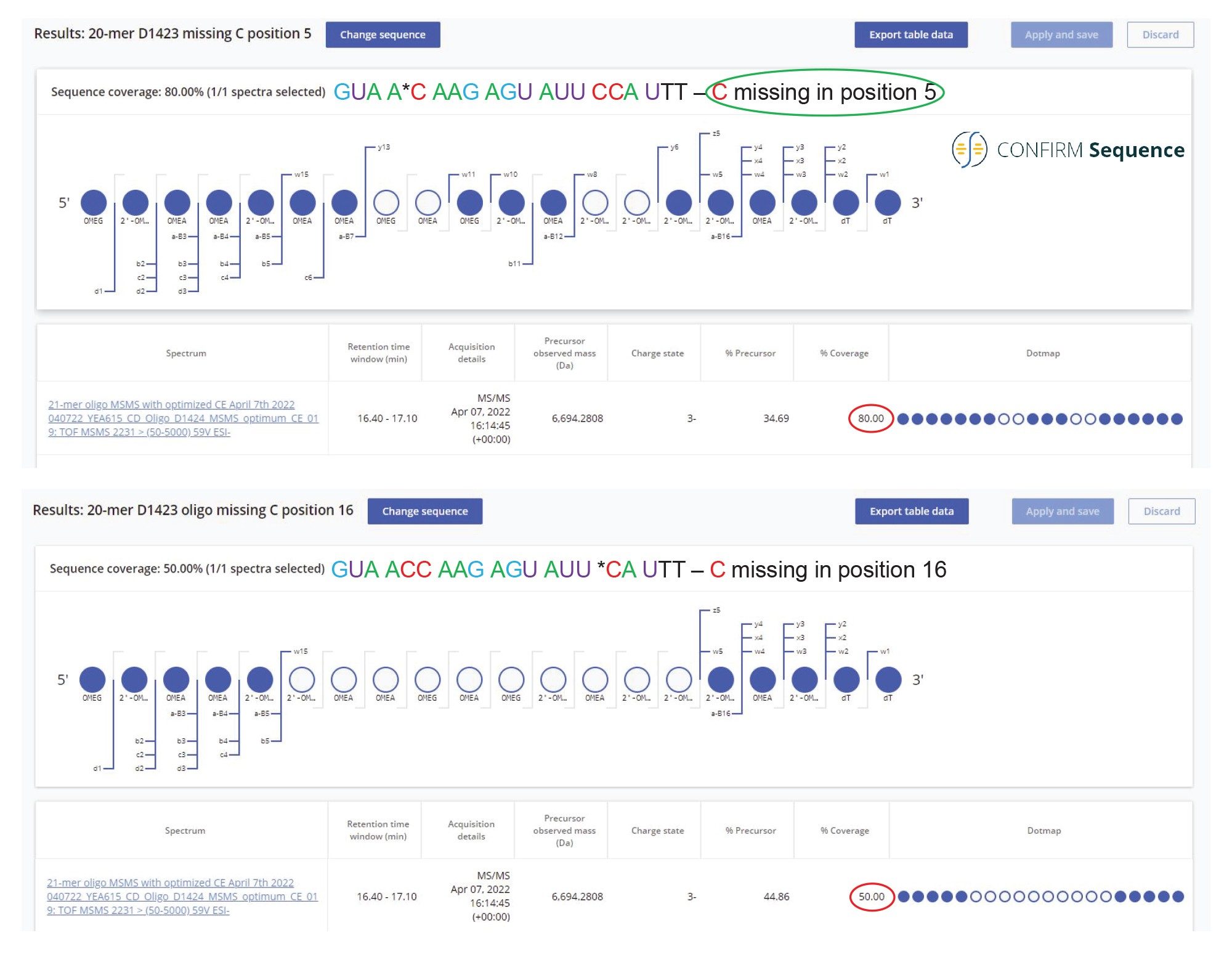 Figure 9. Confirm Sequence screenshot showing the sequence coverage of a 20-mer oligonucleotide impurity belonging to the peak doublet 10 shown in Figure 2. The most abundant isomer of this impurity, eluting just before the FLP, was identified as an oligonucleotide missing a 2’-OMe 5 Me cytidine residue in position 5. This oligonucleotide sequence displayed a significantly higher sequence coverage (80%) than an alternative sequence which is missing the same residue at position 16. There are 4 modified cytidines in the 21-mer sequence, but because they are located as pairs, there are only two possible 20-mer impurities missing a single 2’-OMe 5 Me cytidine. The software looked for a missed 2’-OMe 5-Me cytidine from the sequence of the 21-mer and found strong evidence (80% sequence coverage) for assigning this impurity to the sequence: GUA A*C AAG AGU AUU CCA UTT.
Figure 9. Confirm Sequence screenshot showing the sequence coverage of a 20-mer oligonucleotide impurity belonging to the peak doublet 10 shown in Figure 2. The most abundant isomer of this impurity, eluting just before the FLP, was identified as an oligonucleotide missing a 2’-OMe 5 Me cytidine residue in position 5. This oligonucleotide sequence displayed a significantly higher sequence coverage (80%) than an alternative sequence which is missing the same residue at position 16. There are 4 modified cytidines in the 21-mer sequence, but because they are located as pairs, there are only two possible 20-mer impurities missing a single 2’-OMe 5 Me cytidine. The software looked for a missed 2’-OMe 5-Me cytidine from the sequence of the 21-mer and found strong evidence (80% sequence coverage) for assigning this impurity to the sequence: GUA A*C AAG AGU AUU CCA UTT.
![MS/MS spectrum recorded for the most abundant isomer of the 21-mer oligonucleotide impurity labeled as peak 10 and eluting just before the FLP. The [M-3H]-3 precursor of this oligo (m/z = 2231.0) was fragmented using an optimized fixed collision energy (set at 59 V) in the collision cell of a Xevo G2-XS QTof instrument. The a-B5 fragment ion detected at m/z=1506.29 confirms the presence of one 2’-OMe 5-Me cytidine close to the 5’-end of this oligo, while the w10 fragment ion present at m/z= 1656.77 indicates that the other two 2’-OMe 5-Me cytidines are located at the other end of the molecule](/content/dam/waters/en/app-notes/2022/720007677/720007677en-f10.jpg.82.resize/img.jpg) Figure 10. MS/MS spectrum recorded for the most abundant isomer of the 21-mer oligonucleotide impurity labeled as peak 10 and eluting just before the FLP. The [M-3H]-3 precursor of this oligo (m/z = 2231.0) was fragmented using an optimized fixed collision energy (set at 59 V) in the collision cell of a Xevo G2-XS QTof instrument. The a-B5 fragment ion detected at m/z=1506.29 confirms the presence of one 2’-OMe 5-Me cytidine close to the 5’-end of this oligo, while the w10 fragment ion present at m/z= 1656.77 indicates that the other two 2’-OMe 5-Me cytidines are located at the other end of the molecule.
Figure 10. MS/MS spectrum recorded for the most abundant isomer of the 21-mer oligonucleotide impurity labeled as peak 10 and eluting just before the FLP. The [M-3H]-3 precursor of this oligo (m/z = 2231.0) was fragmented using an optimized fixed collision energy (set at 59 V) in the collision cell of a Xevo G2-XS QTof instrument. The a-B5 fragment ion detected at m/z=1506.29 confirms the presence of one 2’-OMe 5-Me cytidine close to the 5’-end of this oligo, while the w10 fragment ion present at m/z= 1656.77 indicates that the other two 2’-OMe 5-Me cytidines are located at the other end of the molecule.
Conclusion
- A novel waters_connect application - CONFIRM Sequence, is introduced for fast interpretation of complex MS/MS and MSE (DIA) oligonucleotide mass spectra.
- The CONFIRM Sequence application is shown to be capable of providing complete sequence coverage (100%) for the full-length-product (FLP) as well as for low-abundance oligonucleotide impurities, as low as 0.2% abundance levels according to UV peak area measurements.
- Data-independent acquisition (MSE) acquired for the FLP with an appropriate collision energy ramp provides complete sequence coverage without the need to optimize the optimization of individual oligonucleotide precursors.
- The CONFIRM Sequence app is capable of finding sequence omissions, insertions or sequence variants derived from the FLP sequence.
References
- Sharma VK, Watts JK Oligonucleotide therapeutics: chemistry, delivery and clinical progress, Future Med Chem, 2015, 7(16), 2221-2242.
- Roberts TK, Langer R, Wood MJA Advances in oligonucleotide drug delivery, Nat Reviews, 2020, 19, 673-694.
- Pourshahian S Therapeutic oligonucleotides, impurities, degradants, and their characterization by mass spectrometry, Mass Spectrometry Reviews, 2019, 00, 1–35.
- Obika S, Sekine M – editors of Synthesis of therapeutic oligonucleotides, 1st edition, Springer, 2018.
- Capaldi D, Teasdale A, Henry S, Akhtar N, den Besten C, Gao-Sheridan S, Kretschmer M, Sharpe N, Andrews B, Burm B, Foy J Impurities in oligonucleotide drug substances and drug products, impurities, degradants, and their characterization by mass spectrometry, Nucleic Acid Ther, 2017, 27, 309–322.
- An Automated Compliance-Ready LC-MS Workflow for Intact Mass Confirmation and Purity Analysis of Oligonucleotides, Waters application note, 720006820, 2020.
- Intact Mass Confirmation Analysis on the BioAccord LC-MS System for a Variety of Extensively Modified Oligonucleotides, Waters application note, 720007028, 2020.
- Analysis of Oligonucleotide Impurities on the BioAccord System with ACQUITY Premier, Waters application note, 720007301, 2021.
- LC-MS Analysis of siRNA, Single Guide RNA and Impurities Using the BioAccord System with ACQUITY Premier and New Automated INTACT Mass Application, Waters application note, 720007546, 2022.
- McLukey SA, Van Berkel GJ, Glish GL Tandem Mass Spectrometry of Small, Multiply Charged Oligonucleotides, J Am Soc Mass Spectrom, 1992, 3, 60–70.
- Rozenski J, McCloskey J SOS: A simple interactive program for ab initio oligonucleotide sequencing by mass spectrometry, J Am Soc Mass Spectrom, 2002, 13, 200–203.
- Oberacher H, Parson W, Oefner PJ, Mayr BM, Huber CC Applicability of tandem mass spectrometry to the automated comparative sequencing of long-chain oligonucleotides, J Am Soc Mass Spectrom, 2004, 15, 510–522.
- Kretschmer M, Lavine G, McArdle J, Kuchimanchi S, Murugaiah V, Manoharan M An automated algorithm for sequence confirmation of chemically modified oligonucleotides by tandem mass spectrometry, Anal Biochem, 2010, 405, 213–223.
- Nakayama H, Akiyama M, Taoka M, Yamauchi Y, Nobe Y, Ishikawa H, Takahashi N, Isobe T Ariadne: a database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data Nucleic Acids Res, 2009, 37, 1–13.
- Yang J, Leopold P, Helmy R, Parish C, Arvary B, Mao B, Meng F Design and application of an easy to use oligonucleotide mass calculation program, J Am Soc Mass Spectrom, 2013, 24, 1315–1318.
- Nyakas A, Blum LC, Stucki SR, Reymond JL, Schurch S OMA and OPA – software -supported mass spectra analysis of native and modified nucleic acids, J Am Soc Mass Spectrom, 2013, 24, 249–256.
- Rozenski J Mongo oligo mass calculator. Available at URL: http://rna.rega.kuleuven.be/masspec/mongo.htm. Accessed June 22, 2022.
- Kass, I Spectrum Tools, 2018.
- Rentel C, DaCosta J, Roussis S, Chan J, Capaldi D, Mai B Determination of oligonucleotide deamination by high resolution mass spectrometry, J Pharm Biomed Anal, 2019, 173, 56-61.
Featured Products
720007677, July 2022