Routine Determination of Trifluoroacetic Acid (TFA) and Difluoroacetic Acid (DFA) in Surface and Drinking Water by Direct Injection Using UPLC™-MS/MS
Abstract
The purpose of this work is to demonstrate a direct injection UPLC-MS/MS method for the determination of TFA and DFA in surface and drinking water. The method performance study was completed on an ACQUITY™ UPLC I-Class PLUS System with a Xevo™ TQ-XS and Electrospray ion source combination using a Waters™ Atlantis™ Premier BEH C18 AX Column. Samples were prepared by pipetting into a polypropylene injection vial and adding internal standard.
The main method performance was carried out on three common sources of drinking water including tap water from known soft and hard water areas and bottled mineral water, and two types of environmental surface water sampled from a reservoir and a river. The assessment was made by fortifying samples with 300, 500, and 1000 ng/L, in addition to incurred residue concentrations, with three replicates at each level. Internal standards were used for both compounds to monitor and compensate for any variability. Average method performance for trueness was 80 to 110% across all matrices. RSDs were at or within 9% for all compounds with one exception of the low spike level for hard water which was within 25% RSD.
Calibration graph residual values were within 20% and coefficient of determination R2 values above 0.9995 using a linear regression fit at 1/X weighting. Retention time stability tested with over 200 consecutive injections of a soft tap water matrix standard was demonstrated with a RSD below 0.9%, and below 3.0% RSD across all matrix types throughout the assessment.
Benefits
- Limited sample preparation of small samples volumes to speed up analysis time, enhance sample throughput, and limit sources of sample contamination
- Sensitive analysis to determine residues at concentrations as low as 10 ng/L for detection and quantification of both TFA and DFA in a sevenminute run by UPLC-MS/MS
- A robust and reliable solution for monitoring DFA and TFA in drinking and environmental surface water matrices
Introduction
Due to the increasing and more stringent monitoring and restriction needs within regulations in water for per- and polyfluoroalkyl substances, there has been interest in shorter-chain alternatives to the more well publicised longer-chain PFAS. TFA and DFA are considered ultra-short-chain PFAS, which are defined by a single carbon fluorinated with two or three fluorenes, and they are often overlooked when it comes to PFAS analysis.
TFA is found worldwide in salt and fresh water in its deprotonated form, where it is highly stable and easily mobile. TFA can been formed naturally and travel vast distances by wind and tends to favor entering the aqueous environment due to its poor retention in soil. Further and ever-increasing amounts of TFA are also entering bodies of water through many artificial sources whether directly from industrial wastewater and its treatment, or as breakdown products from one of countless substances when appropriate trifluoromethyl (-CF3) containing precursors are present,1 with potential formation sources from pesticides, pharmaceuticals, and refrigerants.
TFA has the potential to accumulate in terminal water bodies and plants leading to potential risks for aquatic organisms and increased human external exposure through drinking water. Due to the high polarity and water solubility of these substances, the potential for bioaccumulation in humans is considered low, as has been shown for the short-chain perfluorobutane sulfonic acid (PFBS),2 however the presence of these compounds has still been detected in humans.
Recently the German Federal Environment Agency (UBA) issued a revised guidance value for TFA in drinking water (Trifluoressigsäure (TFA)–Gewässerschutz im Spannungsfeld von toxikologischem Leitwert, Trinkwasserhygiene und Eintragsminimierung. 20. Oktober 2020. Umweltbundesamt), based on improved toxicological studies, setting a drinking water health guidance value of 60 μg/L and a target value of 10 μg/L (60,000 ng/L and 10,000 ng/L, respectively). This toxicologically justified value was based on the life-long tolerable daily intake of TFA, in which no harm to human health is to be expected. This guideline replaces the health orientation value (GOW) of 3 µg/L (maximum value that applies if the toxicological data is incomplete)3 and as a result extended monitoring programs at rivers and streams have been initiated to identify potential TFA dischargers (https://www.sciencedirect.com/topics/earth-and-planetary-sciences/dischargers) and emission routes responsible for high concentrations.
The analytical determination of these compounds is challenging due to their high polarity, resulting in low retention using reversed-phase liquid chromatography2 and the assorted combinations and concentrations of naturally occurring ions in real water samples from varying sources can provide further challenges. Another important analytical consideration is avoiding introducing contamination when trying to reach LOQs in the lower ng/L range. Laboratory consumables and reagents were screened prior to use and no contamination of TFA and DFA was identified during the analysis.
This application note demonstrates the performance of the ACQUITY UPLC I-Class PLUS coupled with a Xevo TQ-XS with five aqueous samples across drinking and surface water matrix types. These samples were aliquoted into a polypropylene autosampler vial with internal standard and directly injected into the UPLC-MS/MS without any additional concentration or clean-up.
Experimental
Sample Description
One liter of water was collected from sources of drinking and surface water within the UK and stored in 50 mL centrifuge tubes at room temperature until analysis. Surface water included water sampled from a variety of sources and locations such as streams, rivers, reservoirs, and mineral water was purchased from a UK retail outlet and stored at room temperature in its original container. Further surface and ground water samples from sources in Southeast Germany were kindly provided by the Helmholtz Centre for Environmental Research (UFZ). As required detection limits are in the low ng/L range and TFA contamination from sources such as consumables and reagents is possible, laboratory materials were screened for the presence of DFA and TFA before use in the collection, preparation, and analysis of samples.
Components in the liquid chromatography system can also contribute to contamination when analysing compounds such as PFAS. Therefore, precautions were taken to minimise these contributions, and as such, PEEK solvent lines (p/n: 430002198) comprised of PFAS-free components were installed, which replace the conventional Teflon coated solvent lines. This item is also available as a component within the PFAS kit (p/n: 205000588). The isolator column was not used in this application.
Method Conditions
490 µL water sample was aliquoted directly into a polypropylene autosampler vial (p/n: 186005219) and 10 µL of an isotopically labelled standard containing 2500 ng/L TFA-13C2 (TRC) and DFA-13C2 (Bayer) was added to each. The vial was sealed with a preslit PTFE/silicon septa cap (p/n: 186000305) for direct injection by UPLC-MS/MS. The caps were screened for suitability prior to use.
Blanks and standards were prepared in ultra-pure water dispensed from a Milli-Q system and ranged from 10 to 25000 ng/L for both analytes. Spiked recovery samples were quantified against these calibration standards using internal standard correction and standard addition was also calculated for comparison purposes. Recovery spikes were carried out on five matrices with three replicates at each level fortified with 300 ng/L, 500 ng/L, and 1000 ng/L for each compound, spiked in addition to incurred residue levels. These levels were chosen based on the typical residue levels expected in the samples. Table 1 summarizes the final concentration values for each recovery level taking incurred residues into account.
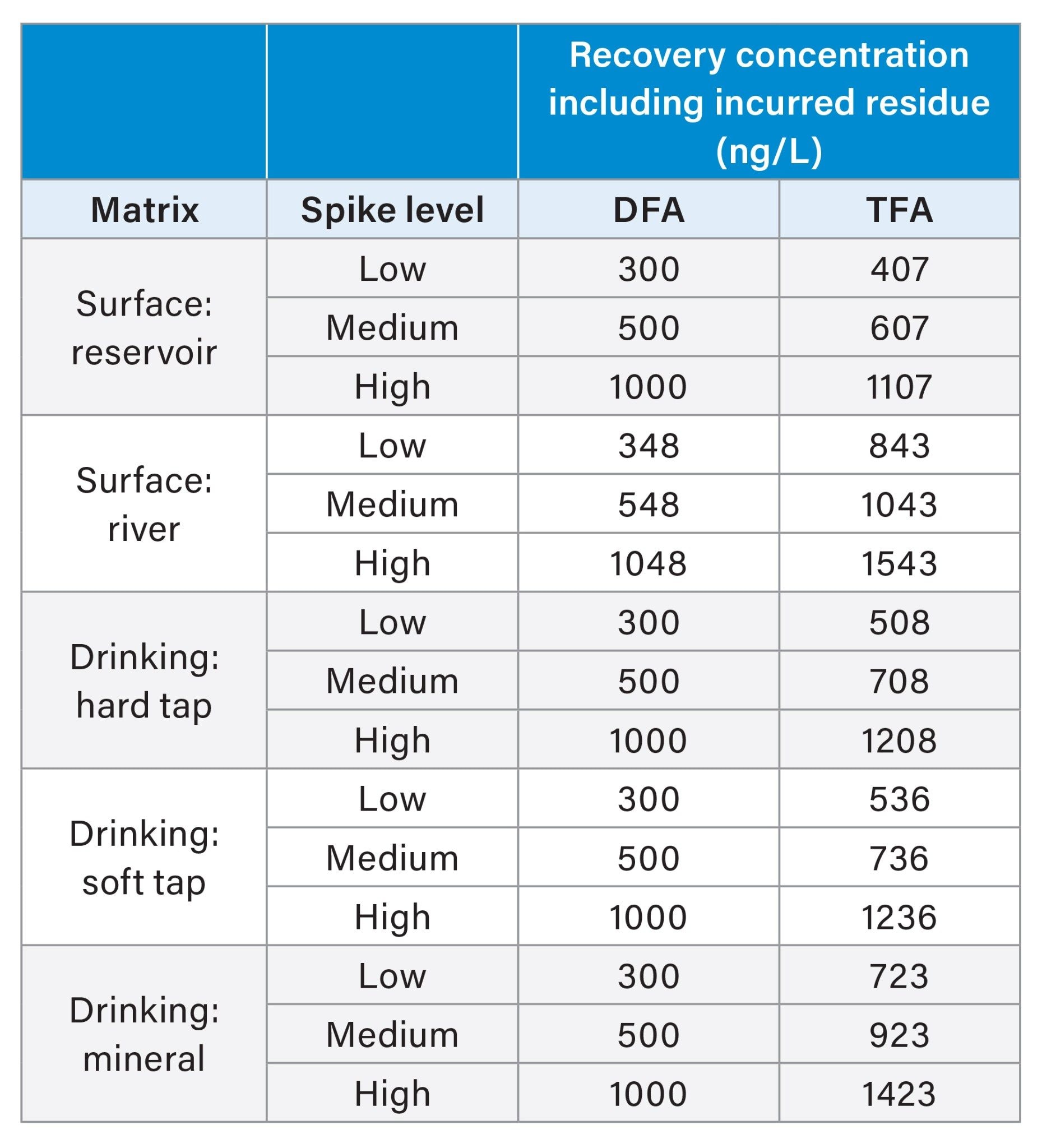 Table 1. Final recovery concentrations for all samples fortified at 300, 500, and 1000 ng/L in addition to incurred residues.
Table 1. Final recovery concentrations for all samples fortified at 300, 500, and 1000 ng/L in addition to incurred residues.
Standard Addition Quantification
Although final reported concentrations were calculated by plotting on a calibration line, residual concentrations can also be quantified automatically by using the standard addition function within the TargetLynx processing method. For this procedure to work effectively, a portion of the sample is analysed as blank, and other portions are spiked with the compound of interest at one or more concentration levels which are appropriate to what concentration is expected in the samples. In the investigation 300, 500 and 1000 ng/L levels were used. When the samples were analysed, a calibration line was obtained, and the concentration was calculated automatically by extrapolating the calibration curve to y=0, with the resulting x value mirrored on the y axis. An example of a calculated TFA residue in soft tap water using the automated standard addition function in TargetLynx is displayed in figure 1.
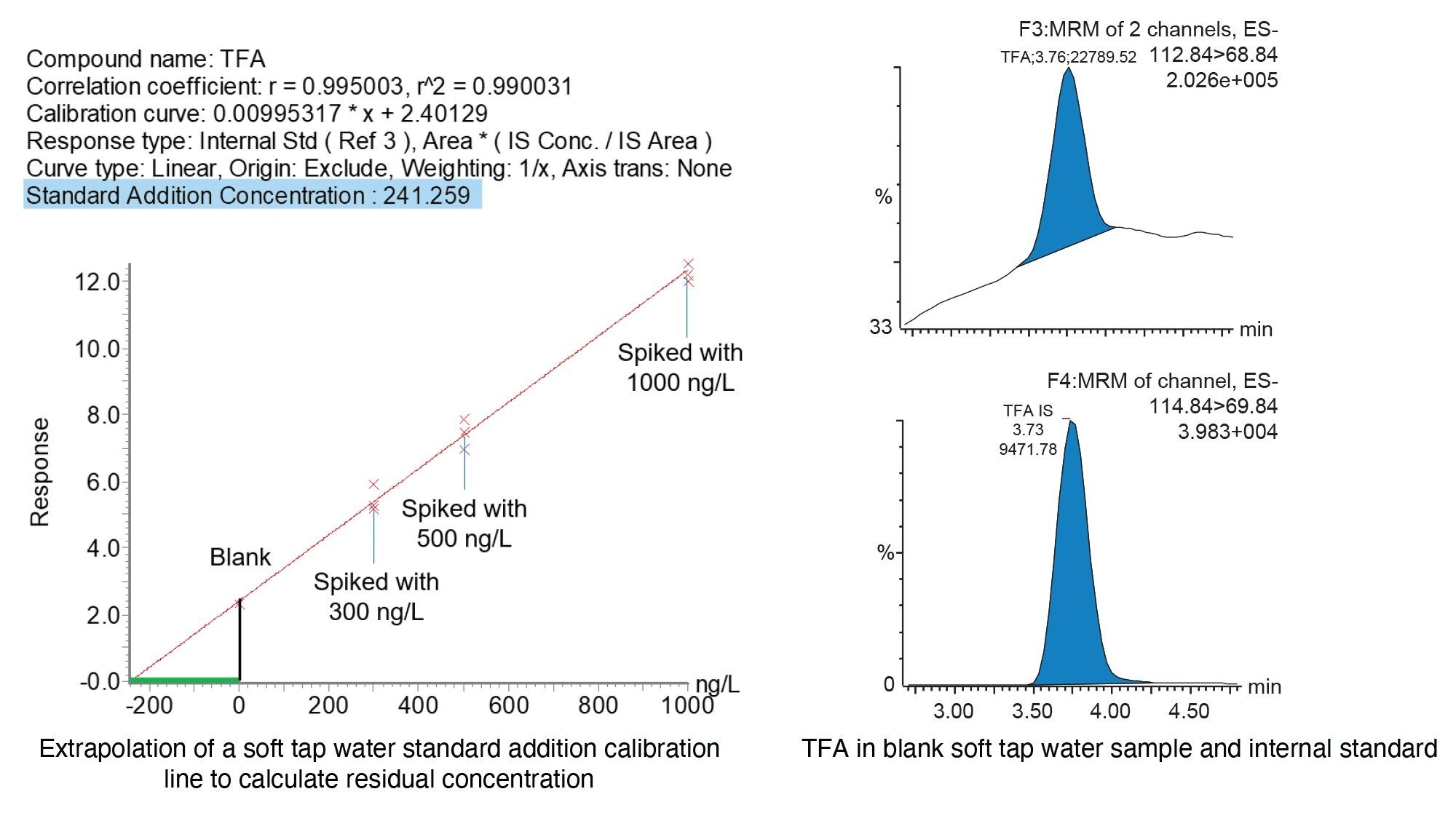 Figure 1. Standard addition function in TargetLynx and chromatography for TFA using a blank soft tap water sample. The green line indicates the extrapolated curve below 0 on the x-axis calculating the incurred residue concentration. Resulting residual concentration is displayed in the header of the calibration curve window (highlighted in blue).
Figure 1. Standard addition function in TargetLynx and chromatography for TFA using a blank soft tap water sample. The green line indicates the extrapolated curve below 0 on the x-axis calculating the incurred residue concentration. Resulting residual concentration is displayed in the header of the calibration curve window (highlighted in blue).
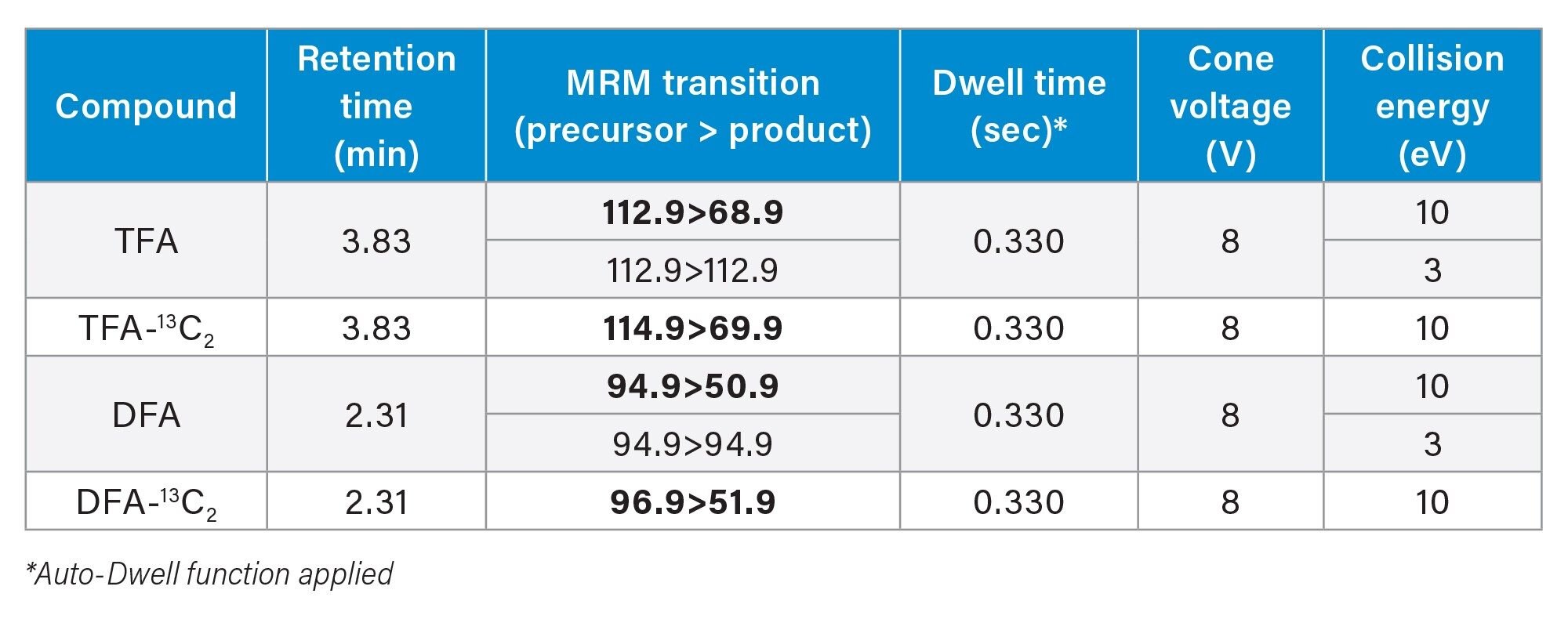
The MRM transitions listed in the MRM transitions table were used in this application for quantification and confirmation of residues. Optimum dwell time for target compounds was set automatically using the auto-dwell function (quantitative transitions are given in bold), so values may vary depending on acquisition windows.
LC Conditions
|
LC system: |
ACQUITY UPLC™ I-Class PLUS with Flow Through Needle (FTN) sample manager fitted with the PFAS kit (p/n: 205000588) |
|
Vials: |
Polypropylene 12 x 32 mm Screw Neck Vial, with Cap and Preslit PTFE/Silicone Septum, 700 µL volume (p/n: 186005221) |
|
Column: |
Atlantis™ Premier BEH C18 AX, 1.7 µm, 2.1 mm X 100 mm (p/n: 186009368) |
|
Column temperature: |
60 °C |
|
Sample temperature: |
15 °C |
|
Injection volume: |
20 µL |
|
Flow rate: |
0.500 mL/min |
|
Mobile phase A: |
2 mM ammonium acetate with 0.001% formic acid |
Gradient Table
MS Conditions
|
MS system: |
Xevo™ TQ-XS |
|
Ionization mode: |
ESI- |
|
Acquisition range: |
MRM |
|
Capillary voltage: |
0.5 kV |
|
Desolvation temperature: |
575 °C |
|
Desolvation gas flow: |
1000 L/hr |
|
Cone gas flow: |
150 L/hr |
|
Source temperature: |
150 °C |
MRM Transitions
Data Management
|
Informatics: |
MassLynx™ v4.2 |
Results and Discussion
When a direct injection approach without sample purification is applied for determination of TFA and DFA in matrix samples, the naturally occurring ions can make challenges in the analysis evident. One of them is signal suppression by coeluting interferences which is corrected using isotope labelled internal standards. Other observable effects were changes in peak shape, variation in the baseline, and changing retention times.
An Atlantis Premier BEH C18 AX Column was used to subjugate these challenges. When operating within the appropriate pH window, the targeted compounds utilize an anion exchange (AX) mechanism in addition to hydrophobic interaction to manipulate selectivity and retention.4 Mobile phase buffer concentration, pH, and organic modifier are three key variables that can be adjusted independently or concurrently, to maximise performance for all analytes. 4
Because of the highly aqueous isocratic mobile phase conditions used in this application, flushing with a neat solvent taking care not to precipitate buffers, is usually sufficient to remove contaminant buildup and prevent the accumulation of organic content on the column. 4 Methanol is recommended for this purpose for when any significant changes in peak shape, peak splitting, shoulders on the peak, shifts in retention, change in resolution, or increasing backpressure may be observed. 4
Correlation between sample pH and retention time
As sensitivity is a challenge at low levels with the omission of an SPE clean-up, a larger starting injection volume of 20 µL was used. With a higher load of sample, the more matrix effects are observed. The peak shape was unaffected by increased injection volume, however for TFA a shift in retention time between real samples and the calibration line was more apparent. The retention time of the internal standard was an effective reference and shifted accurately in relation to the native analyte. As retention time was consistent and stable within matrix and volume of sample injected- this was not considered a result of drift and therefore sample properties were examined as a possible cause.
The pH of water samples tested ranged between 6.9 and 8.0. A direct correlation between the retention time and the extent of the alkalinity of the samples in relation to the LCMS grade water used for calibration standards and mobile phase was observed. The higher the pH of the sample, and the higher the sample injection volume, the higher the retention time shift compared to LCMS grade water. The pH of the LCMS grade water used in development was tested and was found to be slightly acidic at 5.7, exaggerating the effects of the samples which were all pH neutral to slightly alkaline. However, this may be specific to the manufacturer, and lots tested.
Using ultra-purified water from the in house Milli-Q system to make mobile phase and standards narrowed the retention time gap most effectively as it’s pH was found to be close to neutral. The water unit was run for minimum two litres to ensure the water had not been sitting in the system, and the water was dispensed into bottles that had been thoroughly rinsed with LCMS grade methanol and ultra-purified water prior to use. This water was screened prior to use, to ensure that it was free from any residual compounds.
Method Validation Study Results
Calibration standards using ultra-pure water from the Milli-Q system were used for method evaluation and example calibration curves are demonstrated in Figure 3. For both analytes, each calibration graph displayed coefficients of determination above 0.9995 using a 1/X linear regression fit, with individual residuals within 20%. Figure 2 also shows typical chromatography for the compounds at a 10 ng/L concentration level, spiked in ultra-pure water.
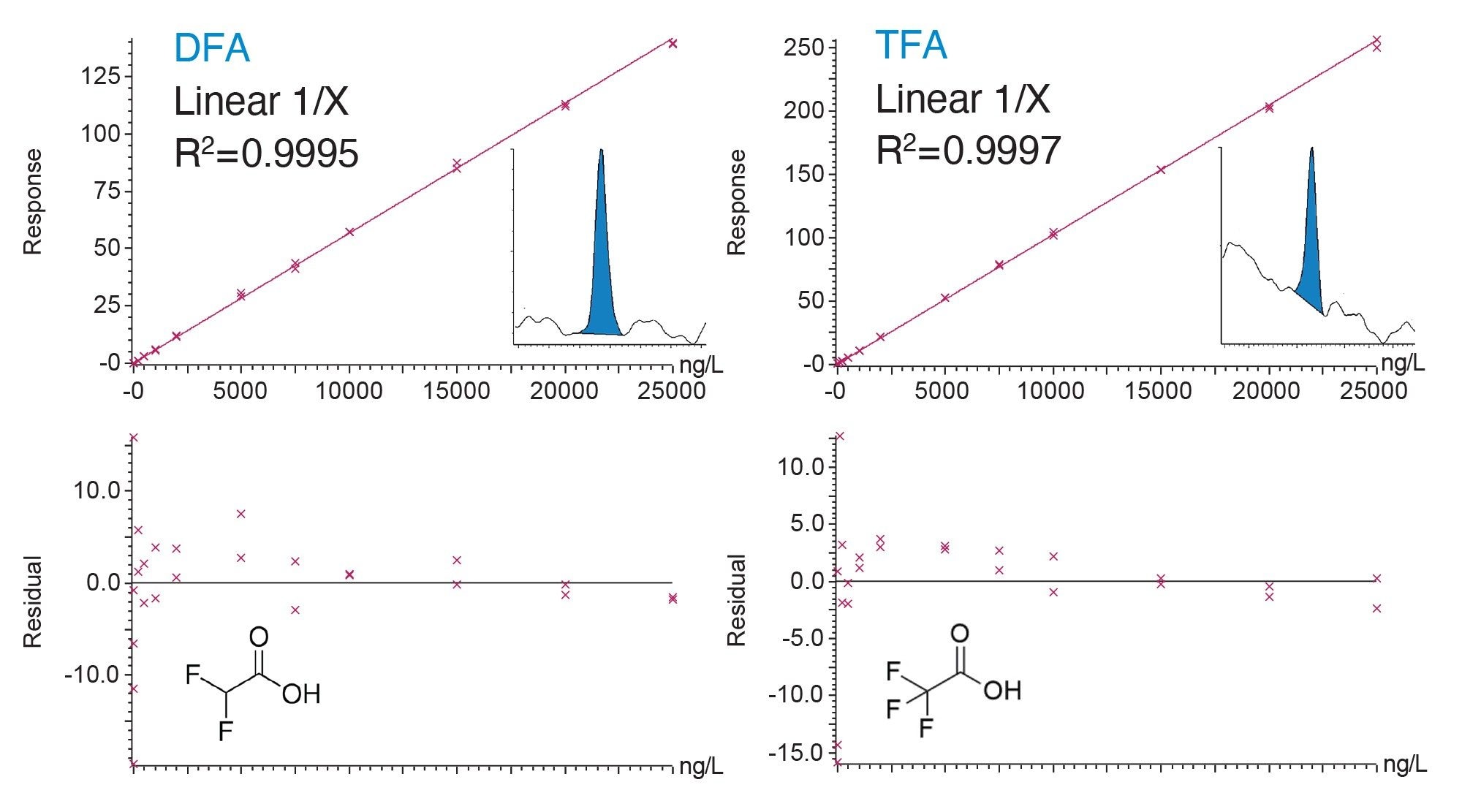 Figure 2. (Top) Bracketed calibration curves for DFA and TFA at 10–25000 ng/L in ultra-pure water including chromatograms for the quantitative transitions at 10 ng/L. All residuals are within 20% of nominal values (bottom).
Figure 2. (Top) Bracketed calibration curves for DFA and TFA at 10–25000 ng/L in ultra-pure water including chromatograms for the quantitative transitions at 10 ng/L. All residuals are within 20% of nominal values (bottom).
Overall, method performance can be summarized in the recovery values highlighted in Figure 3 which covered several typical water types over five samples with a range of properties: each sample was spiked at three levels in addition to incurred residues; 300, 500, and 1000 ng/L. Actual concentrations including incurred residues can be found in Table 1.
All levels and matrices gave an average recovery by compound between 80 and 110% and are summarized in Figure 3. Repeatability of the method was assessed from the recovery samples with most recovery values displaying RSDs below 9% for both compounds. The percentage RSD values are summarized by error bars.
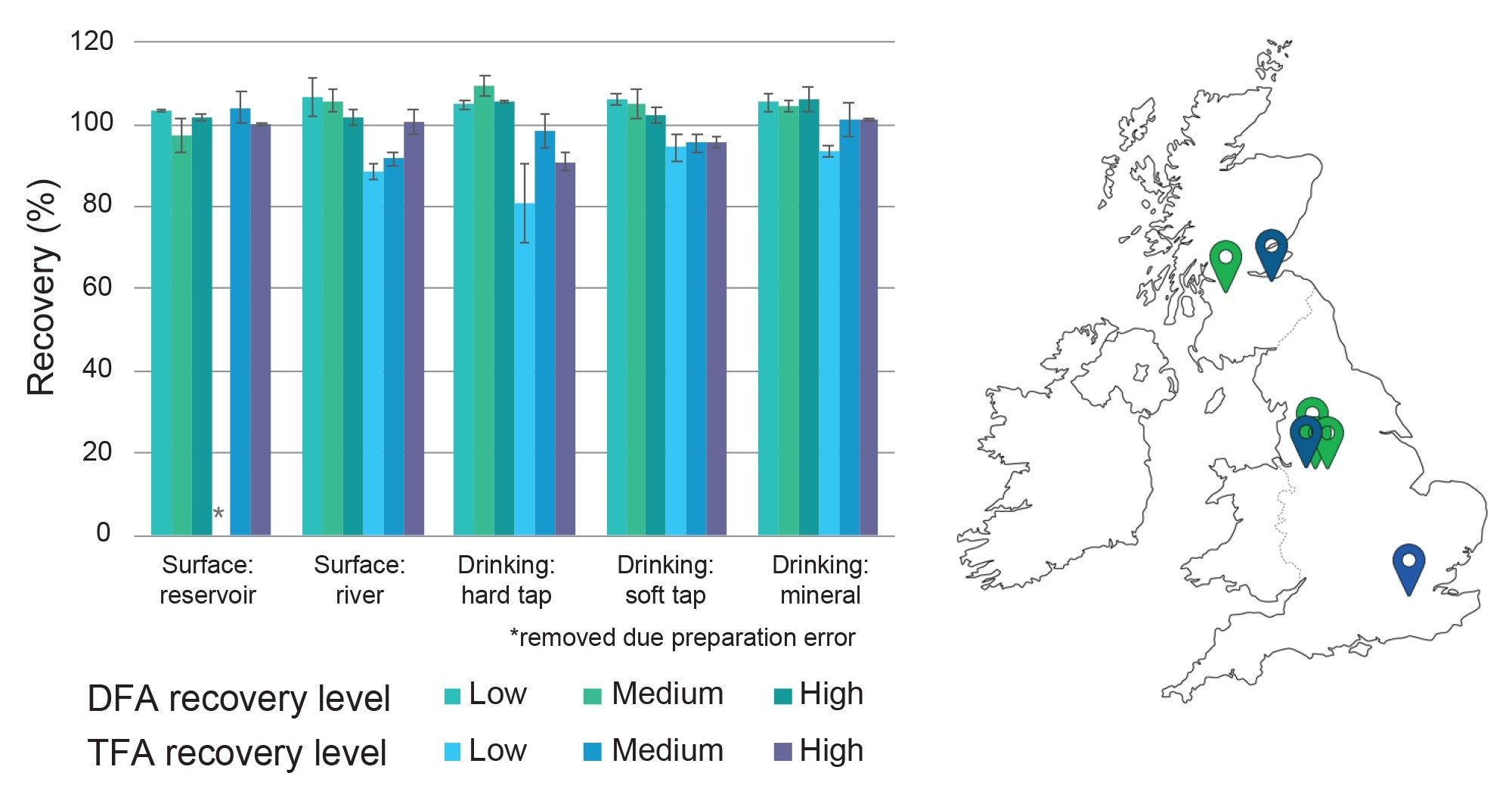 Figure 3. Recovery of DFA and TFA included in method for various water matrix types fortified at 300, 500, and 1000 ng/L in addition to incurred residues. Error bars represent the percentage RSD for each compound across the three data points. Water was collected from various sources around the UK (location tags on map in green and blue indicate surface and tap water sources, respectively).
Figure 3. Recovery of DFA and TFA included in method for various water matrix types fortified at 300, 500, and 1000 ng/L in addition to incurred residues. Error bars represent the percentage RSD for each compound across the three data points. Water was collected from various sources around the UK (location tags on map in green and blue indicate surface and tap water sources, respectively).
The use of an internal standard for DFA was important when quantifying samples relative to the ultra-pure water standards as some of the more complex samples presented a reduction in signal. Although no signal losses were observed for TFA, the use of internal tracked the relative retention time shift between the ultra-pure water standards and matrix samples, with a steady and consistent response within matrix types.
Overall retention time for all analytes across the ultra-pure water, tap and surface water types tested (ten water sources in total, included those donated by the Helmholtz Centre for Environmental Research) was within 3% RSD and demonstrated retention time stability over the study regardless of water type and source. An additional retention time stability test was conducted on a soft tap water sample in which 200 injections of a 200 ng/L matrix calibration standard was run without operator intervention. The sample has been spiked with 200 ng/L DFA only, as there was an endogenous level of approximately 200 ng/L TFA already present in the matrix. Displayed in Figure 4 is a plot of the retention time throughout the course of the run. The retention time was stable throughout and RSDs for both compounds across the whole run were within 0.9% with no significant change to the observed peak shape.
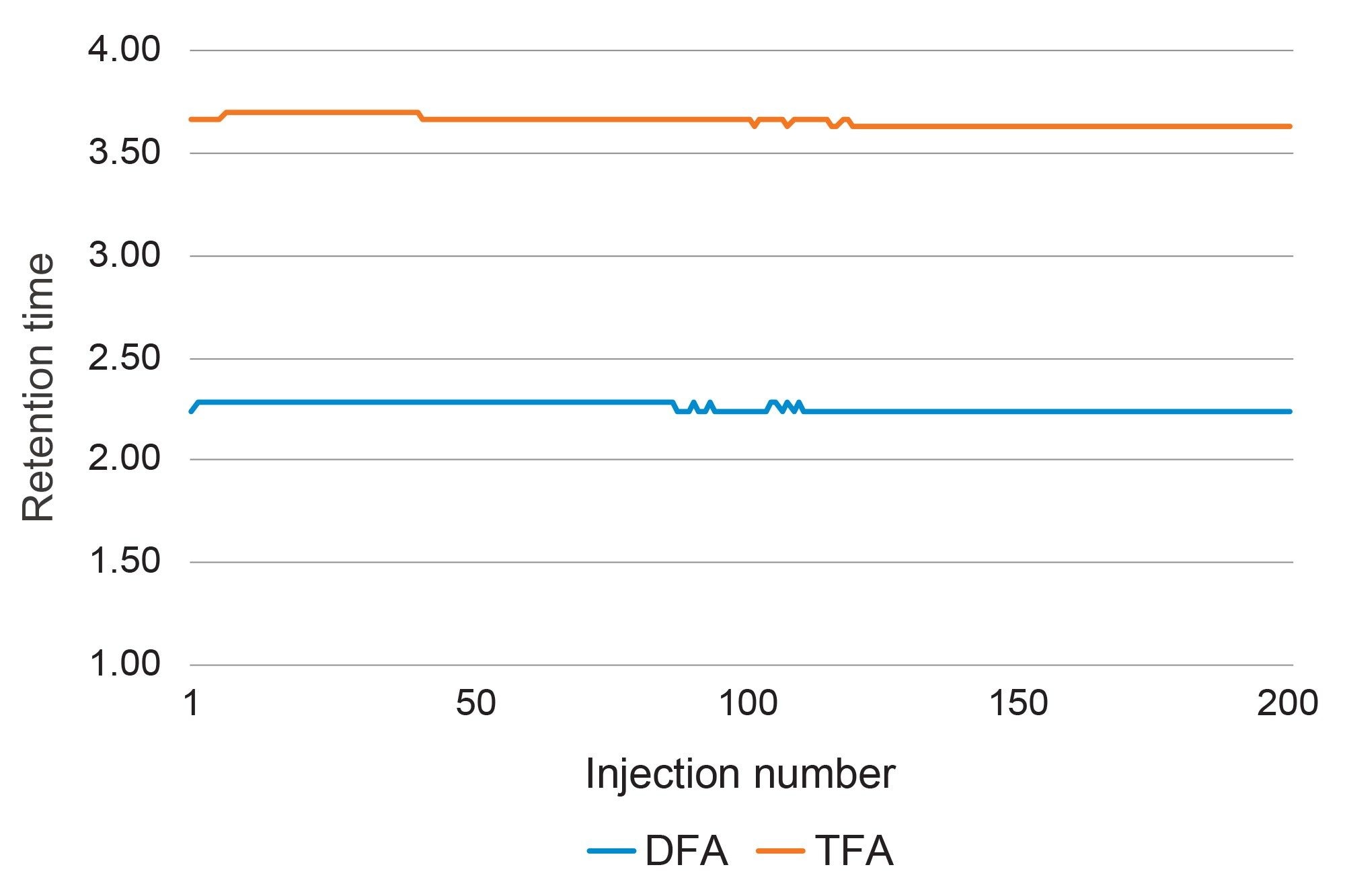 Figure 4. Retention time stability of DFA and TFA across 200 injections of a soft tap water matrix at 200 ng/L. (Percentage RSD values are within 0.9%).
Figure 4. Retention time stability of DFA and TFA across 200 injections of a soft tap water matrix at 200 ng/L. (Percentage RSD values are within 0.9%).
Real-life Water Sample Analysis
Incurred residue sample concentration calculated by standard addition was comparable to those calculated by quantification against the reagent standards using ultra-pure water. Quantification was possible with the use of internal standards for both compounds. All samples with incurred residue concentrations calculated above 10 ng/L by quantitation using an ultra-pure water calibration curve were within 21% of the values calculated by standard addition. A summary of these results can be seen in table 2.
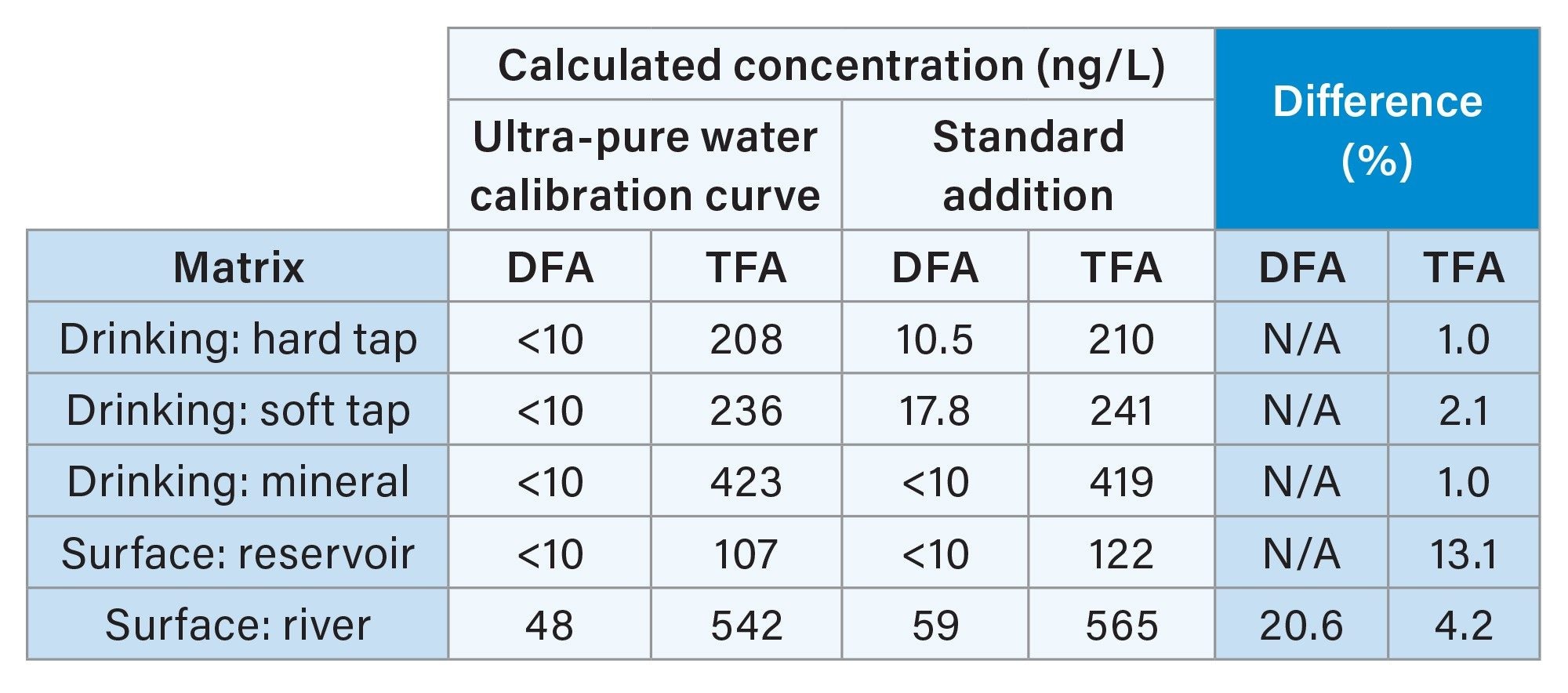 Table 2. Comparison of incurred residue concentrations of samples calculated by quantitation using an ultra-pure water standard calibration curve and standard addition.
Table 2. Comparison of incurred residue concentrations of samples calculated by quantitation using an ultra-pure water standard calibration curve and standard addition.
Through observation of internal standard signal, the response was consistent over all matrix types for TFA. For DFA, river and soft tap water samples displayed a supressed response in relation to reagent standards. The use of an internal standard to correct for this was therefore needed when calculating final concentrations for this compound in these matrices when using an ultra-pure water standard calibration curve.
In addition to the matrices sourced in the UK, surface and groundwater samples collected by the UFZ from sites in Germany were tested, including samples from agricultural backgrounds and those known to contain trace contaminants. Levels of DFA were below the 10 ng/L quantifiable limit, however, levels of TFA present were over ten times the concentrations seen in the highest concentrated samples tested from UK sources, ranging from 7700 to 21000 ng/L.
Conclusion
The method validation study results demonstrate a robust analytical method for the determination of TFA and DFA in drinking and environmental surface water.
A LOQ of 10 ng/L was achieved with a direct injection of sample without the need for additional clean up or concentration steps. Analysis was performed using a Xevo TQ-XS coupled to an ACQUITY UPLC I-Class PLUS and separated using an Atlantis Premier BEH C18 AX Column for accurate and reliable results.
The trueness and precision of this UPLC-MS/MS method determined at three matrix QC levels with three replicate injections for five sample matrices was found to be acceptable with an average recovery between 80 and 110% and peak area RSD values below 9%. This was with the only exception of a spiked low level hard water sample for TFA where the RSD was 19%. Retention time stability and robustness was proven over the course of the study with RSDs for all compounds under 3% across all matrices and samples tested.
Concentrations in samples were successfully calculated for DFA and TFA by quantitation with a 1/X linear calibration line with a range of 10–25000 ng/L, prepared with standards made using ultra-pure water. Incurred sample residues could also be determined by standard addition using the automatic calculation function within TargetLynx. The range of values for incurred residue levels in the samples tested including those donated from the UFZ showed that TFA, and DFA to a lesser extent can be found almost everywhere, highlighting the importance for the need to test and monitor levels in our environment and drinking water supply.
Scientists must validate the method in their own laboratories and demonstrate that the performance is fit for purpose and meets the needs of the relevant analytical control assurance system.
Acknowledgements
Bayer for providing the isotopically labelled standard DFA-13C2 for use in analysis.
Helmholtz Centre for Environmental Research (UFZ) for donating additional surface water samples for testing.
References
- Scheurer. M, Nödler. K, Freeling. F, et al, Small, mobile, persistent: Trifluoroacetate in the Water Cycle – Overlooked Sources, Pathways, and Consequences for Drinking Water Supply, Water Research, Volume 126, 460–471 (2017).
- Björnsdotter. M.K., Yeung, L.W.Y., Kärrman, A. et al. Challenges in the Analytical Determination of Ultra-Short-Chain Perfluoroalkyl Acids and Implications for Environmental and Human Health. Anal Bioanal Chem 412, 4785–4796 (2020).
- EUROPEAN FLUOROCARBONS TECHNICAL COMMITTEE (EFCTC) www.fluorocarbons.org/environment/environmental-impact/tfa-as-an-atmospheric-breakdown-product/. Accessed May 2022.
- Atlantis Premier BEH C18 AX Column care and Use Manual. Waters Manual 720006576EN. 2021 May.
720007765, October 2022