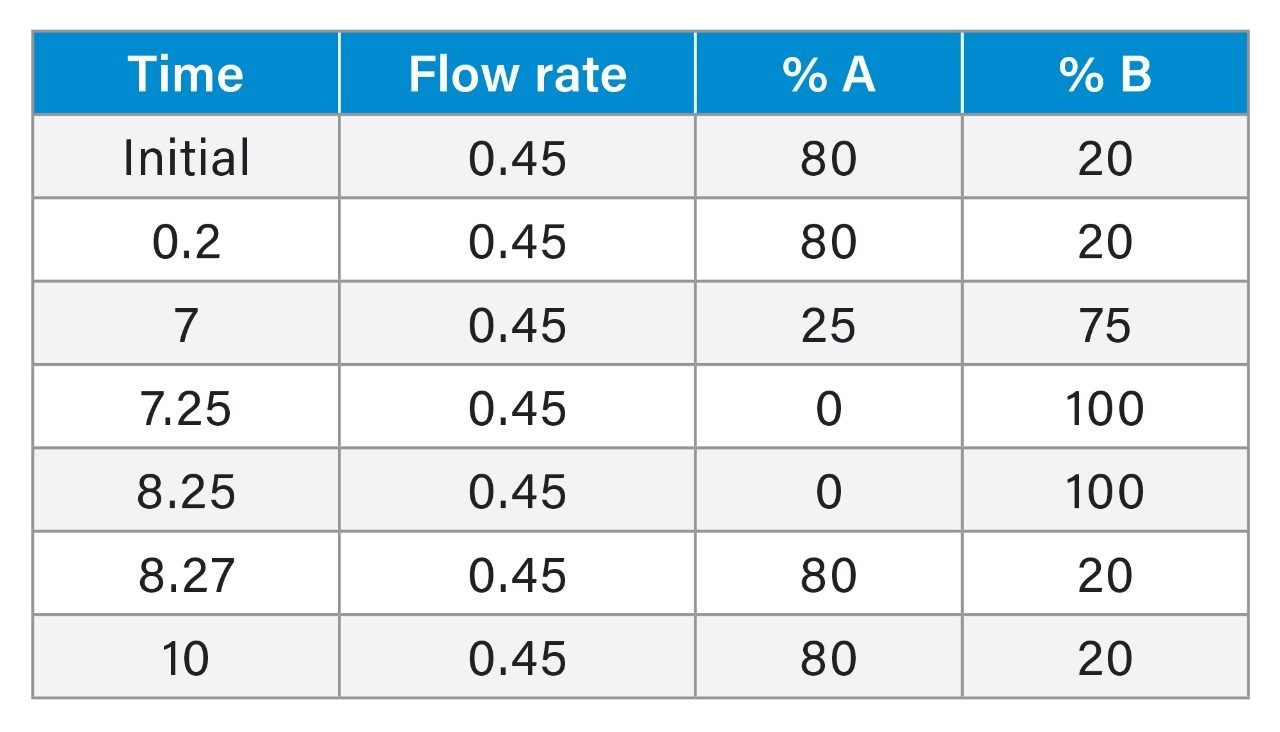
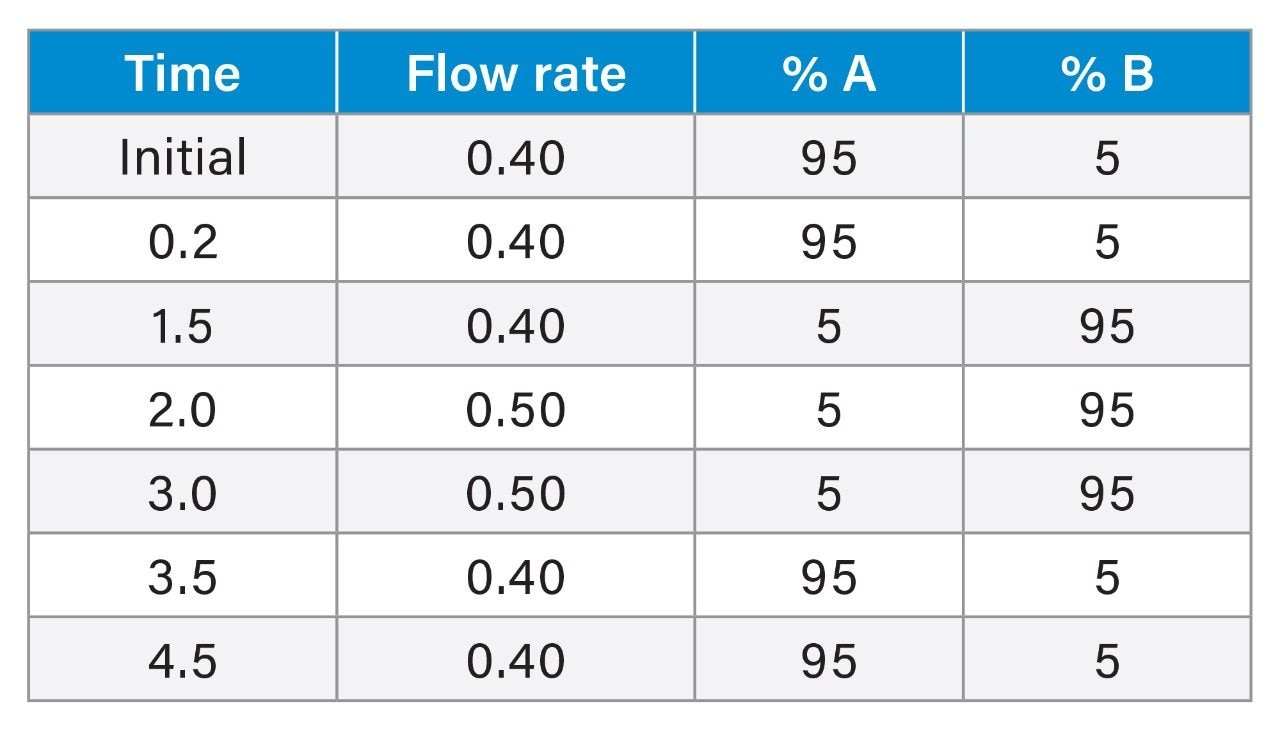
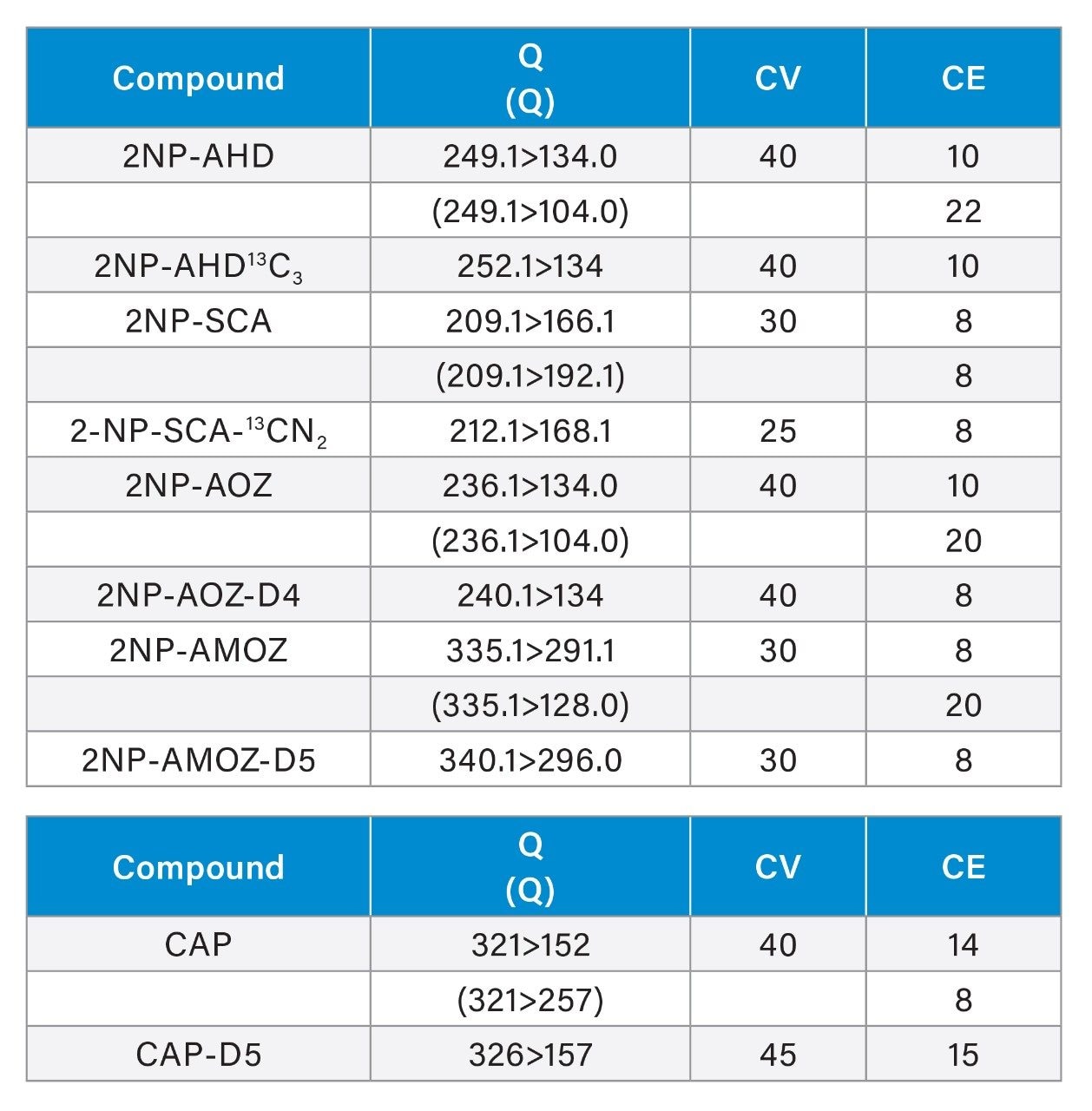
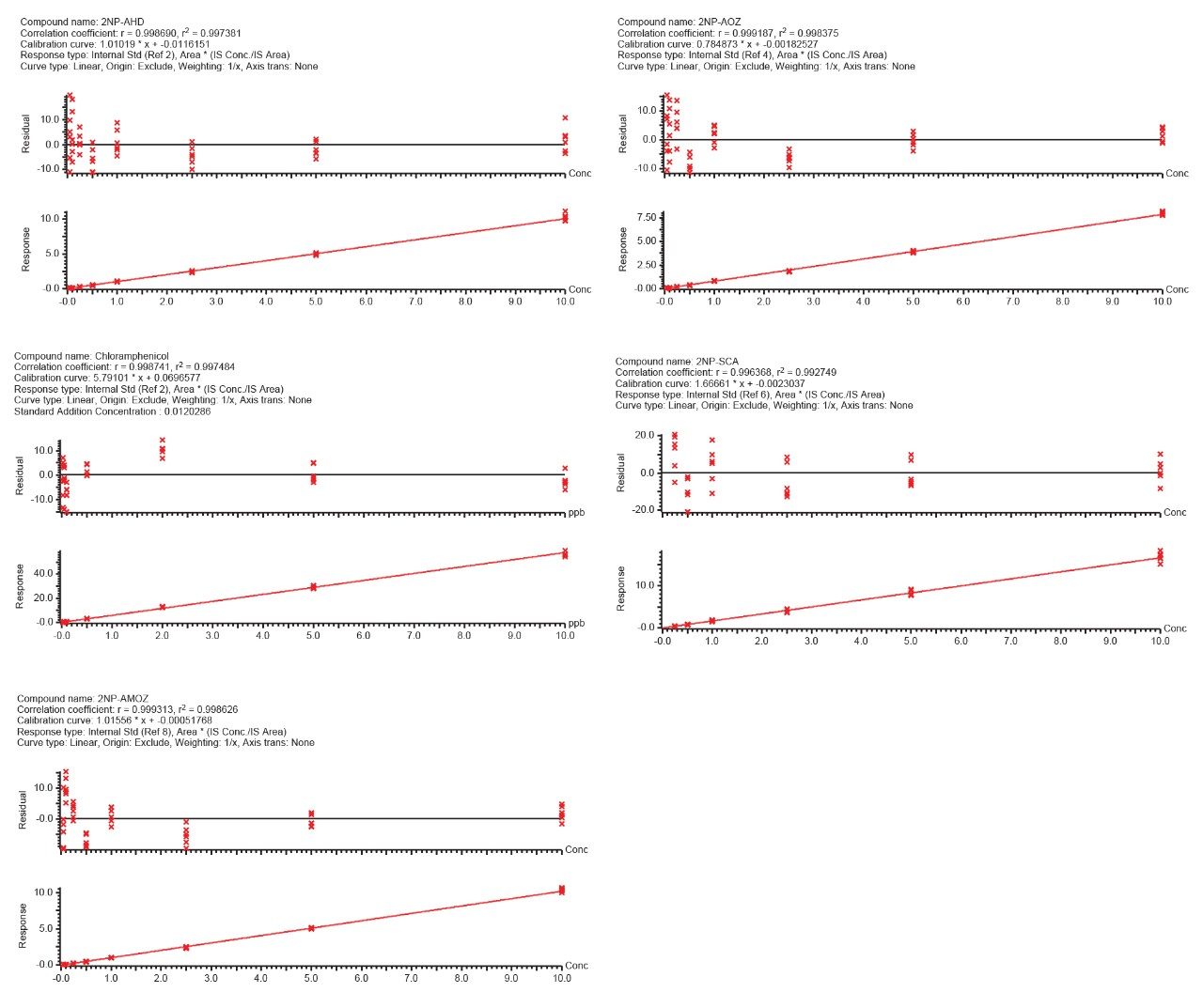
Historically, there has been global concern about the safety of foods of animal origin contaminated with antibacterial residues. Within the European Union, for specific prohibited, or unauthorised pharmacologically active substances. Reference Points for Action (RPAs) in food have been established under Regulation (EU) 2019/1871.1 RPAs are set at the lowest level which can analytically be achieved by the official control laboratories [Regulation (EU) 2017/625].2 By consequence, the RPAs also define the minimum method performance requirements (MMPRs) for these substances in food. Food of animal origin found to contain these residues at concentrations equivalent to, or above the RPA, are considered not to comply with the legislation and shall not enter the food chain.
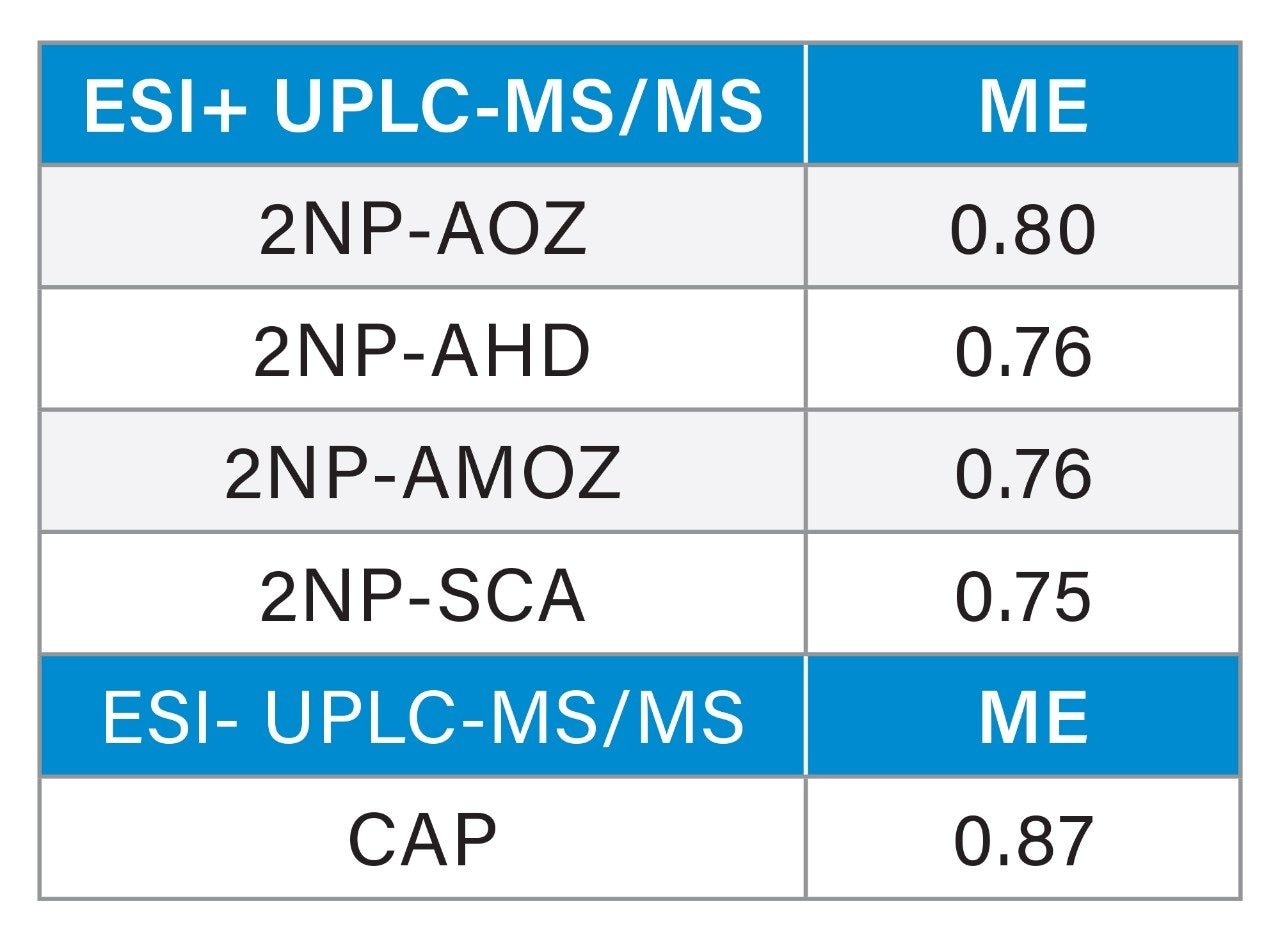
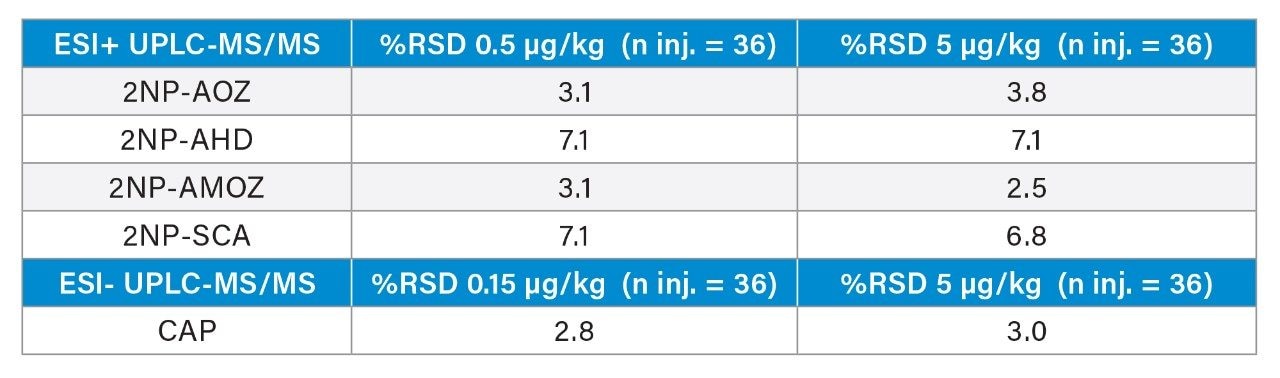
In 2019, RPAs of 0.15, 0.5, and 0.5 µg/kg were established for chloramphenicol, malachite green, nitrofurans, and their metabolites, respectively1 within the EU. The use of these antibacterial compounds in livestock and aquaculture production is also banned in major global geographies by regulations established by the Food and Drug Administration, CODEX Alimentarius, and Joint FAO/WHO Expert Committee on Food Additives (JECFA).3-5
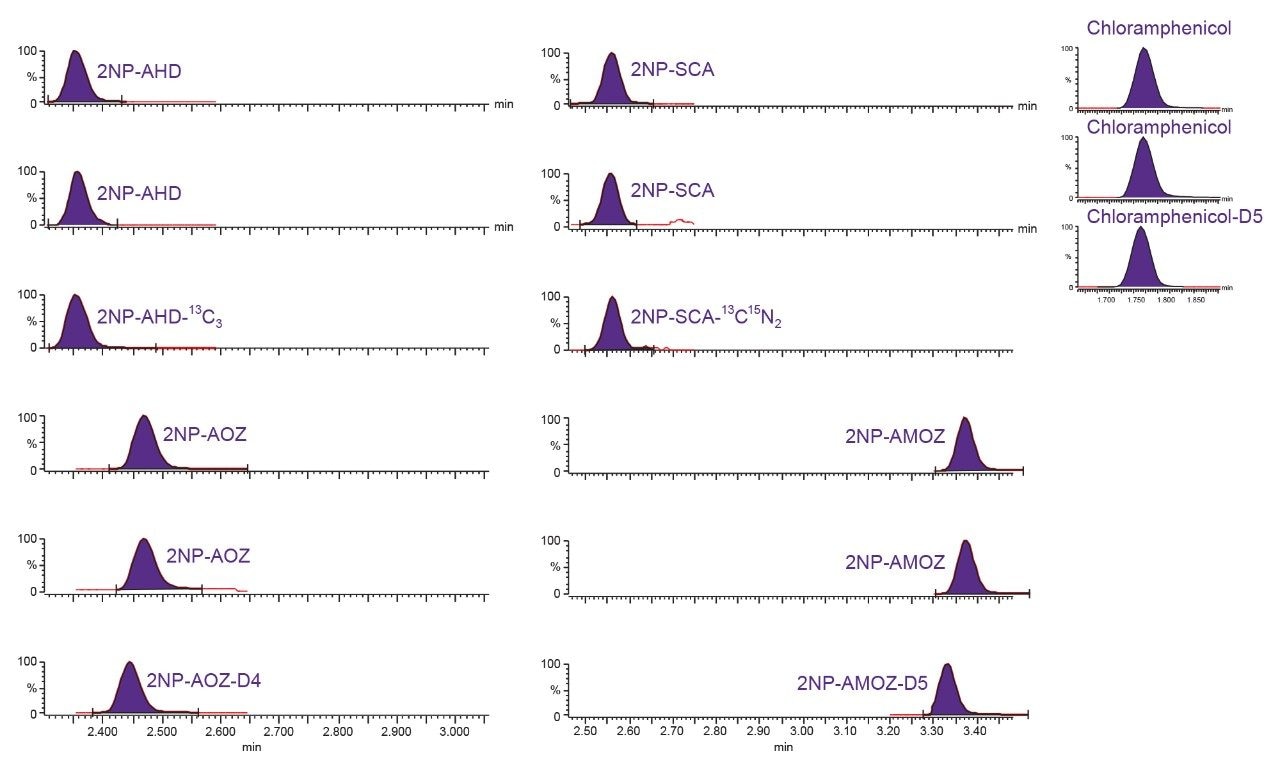
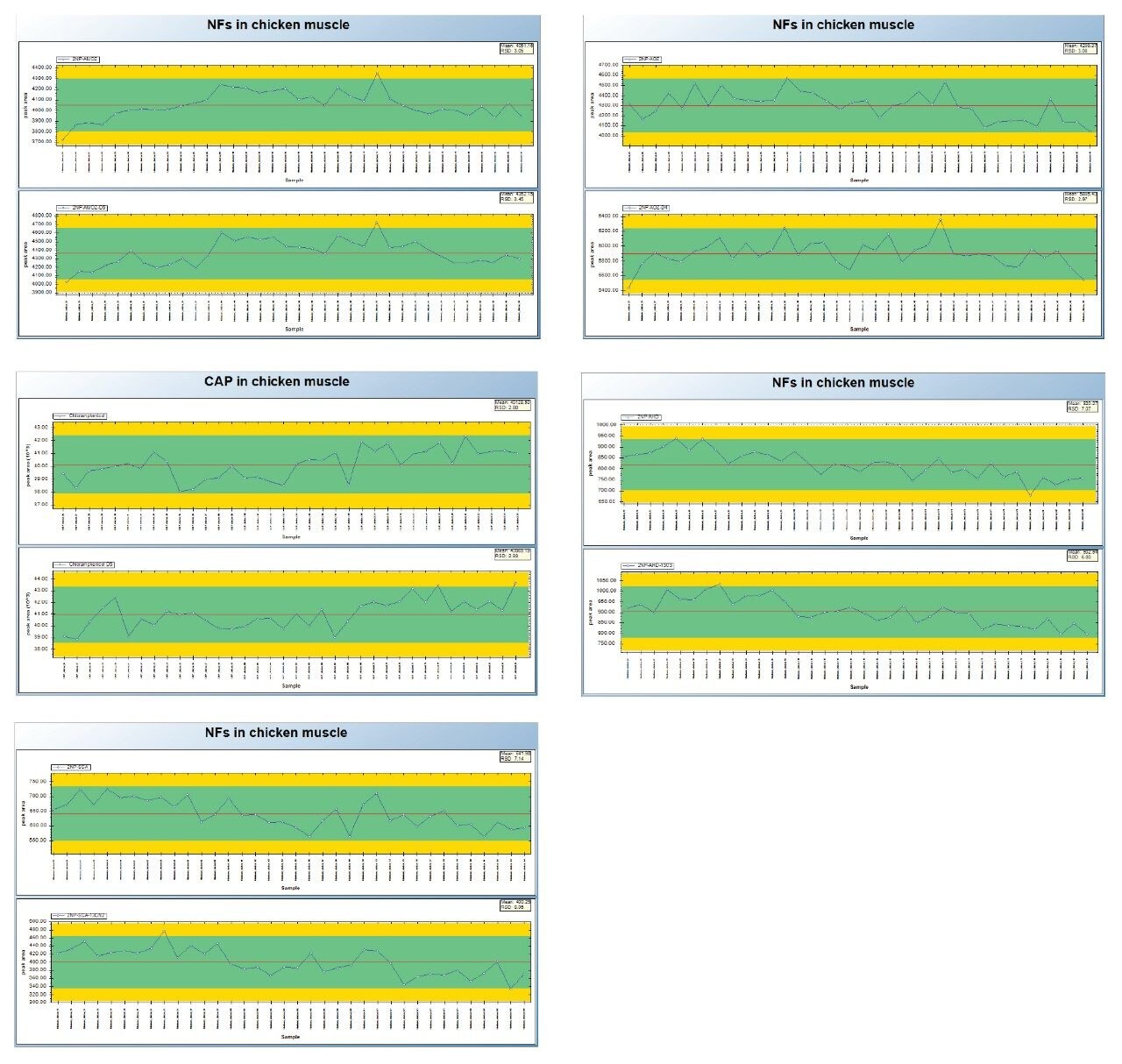
Nitrofurans are metabolised rapidly in vivo but stable tissue-bound metabolites are formed. Fragments of these metabolites may be released by mild acid hydrolysis and monitored as marker residues.6 For example, 3-amino-2-oxazolidinone (AOZ) is monitored as a marker residue for the drug furazolidone. 3-Amino-5-morpholinomethyl-2-oxazolidinone (AMOZ) is the marker residue for furaltadone, and 1-aminohydantoin (AHD) for nitrofurantoin and semicarbazide (SCA) for nitrofurazone. To complete the analysis and present extracts that are amenable to reverse phase chromatography, the resulting metabolites are normally derivatized with 2-nitrobenzaldehyde yielding the ‘NB’ derivatives.
The Xevo TQ-S cronos instrument was developed as a reliable system for robust, routine quantitative analysis in complex matrices. The Xevo TQ-S cronos incorporates a similar sample cone design that has been previously utilized in the extremely popular ACQUITY QDa. In addition to the reverse cone design, established Waters patented technology also contributes towards the robust performance of the Xevo TQ-S cronos including orthogonal geometry ion source, StepWave ion guide, and the T-Wave enabled collision cell. Previously published application notes have reported benefits for other quantitative residue analysis applications including pesticides, acrylamide, and the trimethylphenyl dyes.7-9
This study investigates the performance of the Xevo TQ-S cronos Tandem Quadrupole Mass Spectrometer coupled with the ACQUITY UPLC I-Class PLUS System for the determination of the banned veterinary drug residues of chloramphenicol (CAP) and the nitrofuran metabolites (NF).