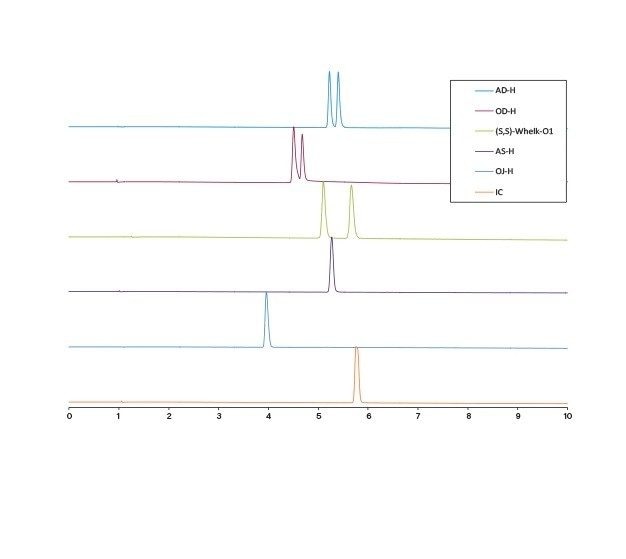
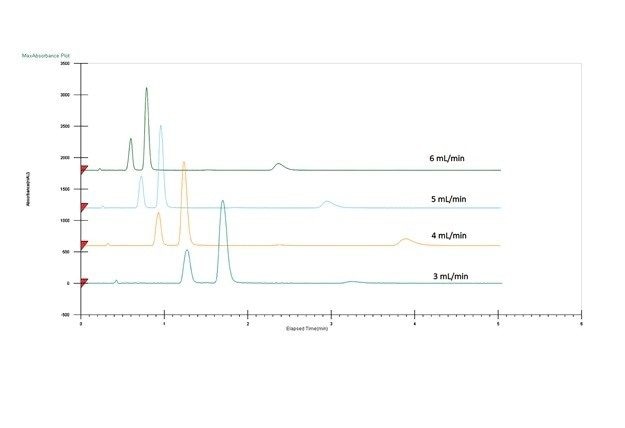
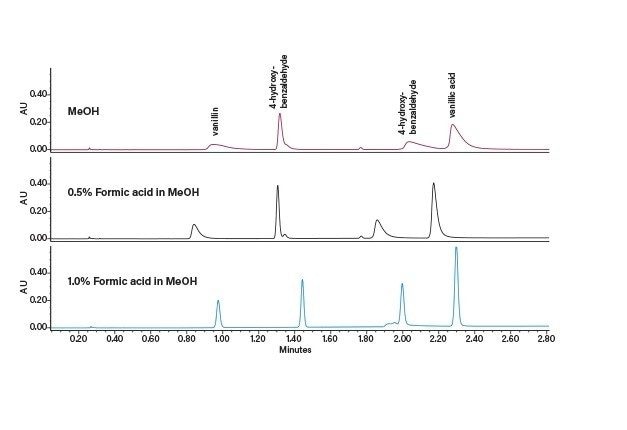
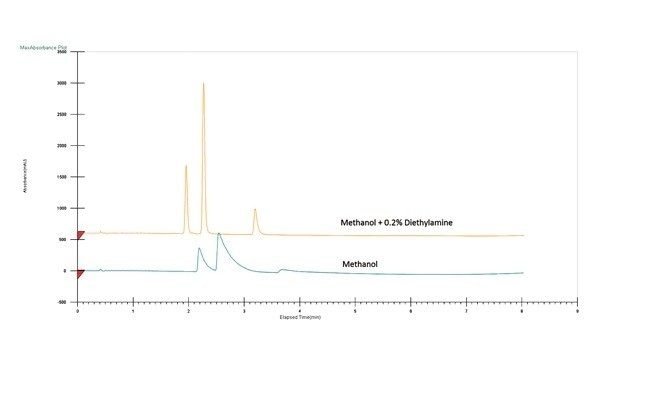
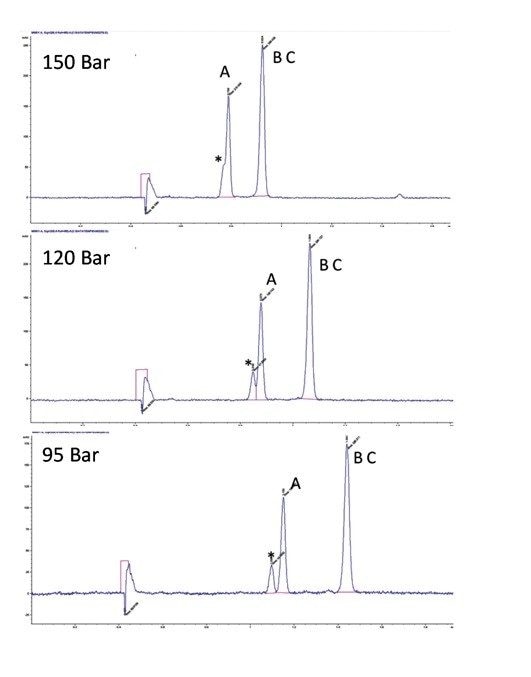
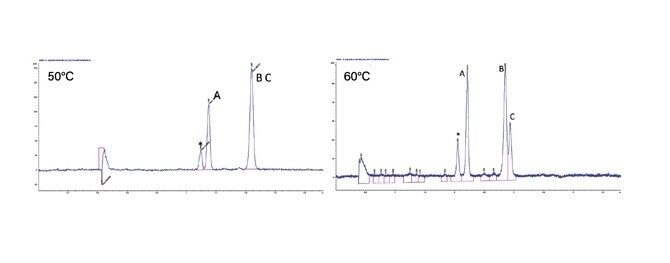
Optimisation des conditions de la phase mobile
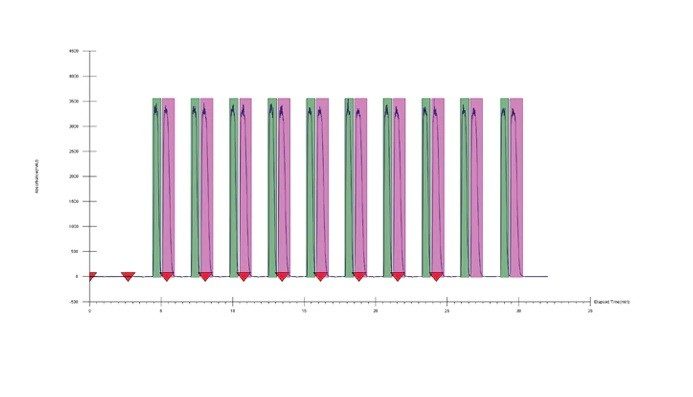
Le choix de la combinaison optimale de solvant et de colonne dépend généralement de l’application ou de l'objectif. Dans un scénario parfait, un chargement élevé, une bonne résolution et une séparation rapide se combinent pour assurer une récupération complète de la cible à une pureté élevée. Mais en réalité, un compromis est généralement nécessaire pour déterminer la solution la plus efficace. Par exemple, les pics peuvent être bien séparés (permettant une charge élevée), mais nécessitent une durée d'analyse plus longue. En revanche, une bonne séparation avec un temps d’analyse significativement plus court permettrait un temps de cycle plus court, mais éventuellement avec un chargement plus faible. Un autre élément à prendre en compte est le traitement en aval ou la récupération des fractions, où la quantité ou le type de solvant peut être un facteur important. Enfin, si la séparation peut être réalisée dans des conditions isocratiques, des injections empilées peuvent être réalisées, ce qui améliore considérablement la productivité. Une fois la colonne et le solvant sélectionnés, d’autres paramètres peuvent être manipulés pour mieux optimiser la séparation avant l’extrapolation et la purification.
Optimisation des conditions de gradient : en SFC, les gradients ciblés ne fonctionnent pas de la même manière qu'en RPLC. En raison des nombreux mécanismes de rétention concurrents en chromatographie en phase normale, des modifications du gradient peuvent avoir des effets disparates sur la rétention de différents analytes. Dans le cas de la SFC en particulier, un changement de densité et une chute de pression accompagnent également l’augmentation du cosolvant à travers le gradient. À mesure que la quantité de cosolvant augmente, l’effet sur le temps de rétention n’est pas linéaire, ce qui rend difficile de prédire la sélectivité des composés en fonction des différentes conditions. Pour les composés structurellement similaires, cela pose moins de problèmes, car ils obéissent à des mécanismes de rétention similaires. Les échantillons contenant des mélanges de composés structurellement différents (dans une matrice, par exemple) posent plus de difficultés. Mais en général, des gradients en pente plus douce entraînent des temps de rétention plus longs et une meilleure résolution.
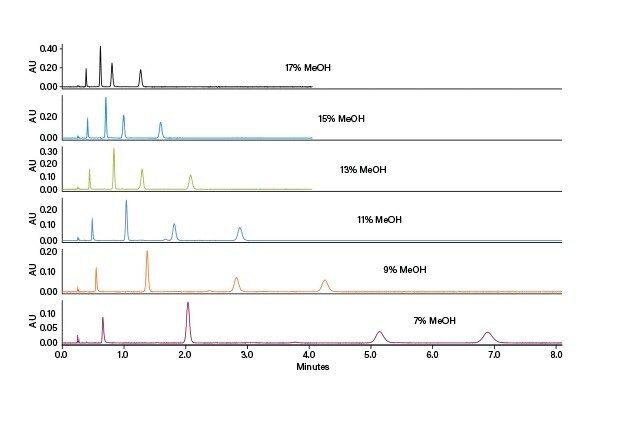
Détermination des conditions isocratiques : les méthodes isocratiques sont idéales, car elles sont faciles à développer sur la base des résultats du criblage, et des injections empilées peuvent être utilisées, ce qui améliore la productivité. Les pourcentages de cosolvant à l’élution peuvent être déterminés grâce au temps de rétention et à la pente du gradient de criblage, et en compensant le délai volumique du système et de la colonne. En SFC, le meilleur point de départ pour l’optimisation se situe généralement 5 % en dessous du pourcentage calculé.
Le gradient de criblage était de 2 à 20 % sur 5 minutes et le retard du gradient était de 0,46 min (déterminé précédemment). Ainsi, avec une pente calculée de 3,6 %/min et un pourcentage de départ de 2 %, le pourcentage de cosolvant à l’élution du premier pic à 4,12 minutes a été calculé à l’aide de l’équation suivante :
■ Pourcentage de cosolvant à l'élution = (temps de rétention – délai de gradient) x pente du gradient + % de départ
■ Pourcentage de cosolvant à l'élution = (4,12 min–0,46 min) x 3,6 %/min + 2 %
■ Pourcentage de cosolvant à l'élution = 15 %
Par conséquent, après soustraction de 5 %, des conditions de cosolvant isocratiques de 10 % ont été utilisées comme point de départ pour l’optimisation. La chromatographie résultante montrait une bonne séparation. Cependant, en augmentant la fraction de cosolvant de la phase mobile à 15 %, les pics étaient toujours bien résolus avec un temps d’analyse plus court. Dans ce cas, la méthode à 10 % permet un chargement plus important, tandis que la méthode à 15 % permet des temps de cycle plus courts.