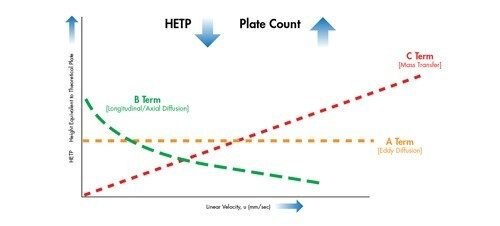
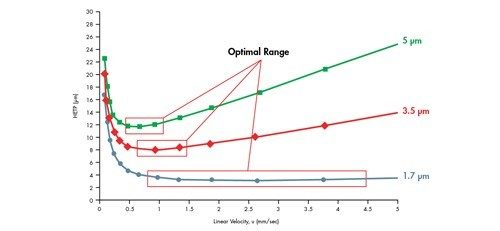
Der A-Term wird als horizontale Linie dargestellt. Er hängt von der Partikelgröße und der Packungsdichte einer Säule ab und ist unabhängig von der Lineargeschwindigkeit [Geschwindigkeit der mobilen Phase]. Mit abnehmender Partikelgröße des Packungsmaterials nimmt auch der H-Wert ab [höhere Effizienz].
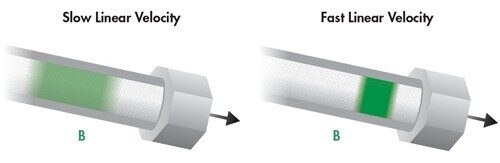
Der B-Term ist als abfallende Kurve mit zunehmender Lineargeschwindigkeit dargestellt. Dieser Term ist unabhängig von der Partikelgröße und gibt an, dass, wenn sich die mobile Phase mit einer langsameren Lineargeschwindigkeit bewegt, die Analytmoleküle länger in der Säule bleiben und daher eine größere Gelegenheit zur Bandenverbreiterung [Diffusion] in Längsrichtung innerhalb der Säule besteht. Umgekehrt bleibt, wenn sich die mobile Phase mit einer höheren Lineargeschwindigkeit bewegt, weniger Zeit für die Diffusion und damit für die Bandenverbreiterung.
Der C-Term wird als zunehmende lineare Beziehung zwischen H und u dargestellt. Unter einer Verteilung von Molekülen treten einige Moleküle in eine Pore der stationären Phase ein, während andere in der beweglichen mobilen Phase bleiben, bis sie ein anderes Partikel erreichen. Darauf folgt der umgekehrte Prozess, bei dem sich immobilisierte Moleküle ablösen und sich im Chromatographiebett weiter nach unten bewegen. Es dauert eine gewisse Zeit, bis sich ein Molekül in die Poren und aus ihnen heraus bewegt hat. Daher wird die Analytbande, die diese Moleküle enthält, über die Länge der Säule verbreitert, wenn Moleküle von einem Partikel zum nächsten übertragen werden. Je kleiner die Partikel der stationären Phase sind, desto schneller läuft dieser Prozess ab und verhindert, dass sich die Analytbande verbreitert. Wenn sich die mobile Phase schnell bewegt, entsteht ein größerer Abstand zwischen den immobilisierten Molekülen und denen, die sich vorwärts bewegen. Dies weist darauf hin, dass sich die mobile Phase mit einer langsameren Lineargeschwindigkeit bewegen sollte, damit die Population der Analytmoleküle zusammenbleibt. Wenn die Geschwindigkeit der mobilen Phase erhöht wird, wird die Population der Analytmoleküle disperser, was zu einer stärkeren Bandenverbreiterung führt.
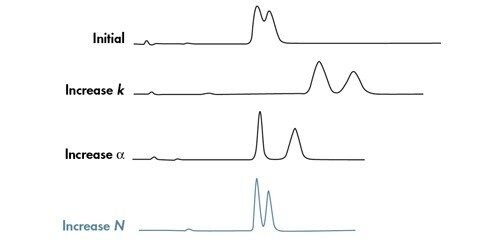
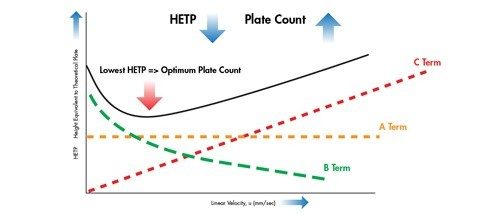
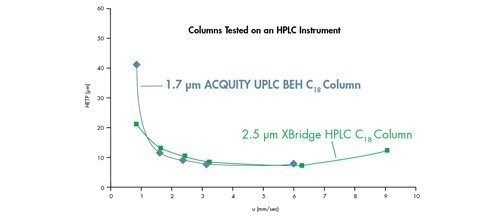
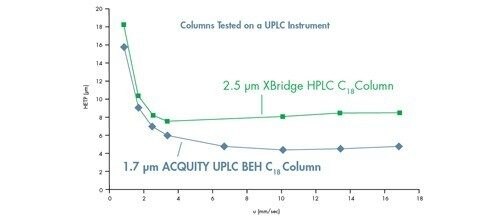
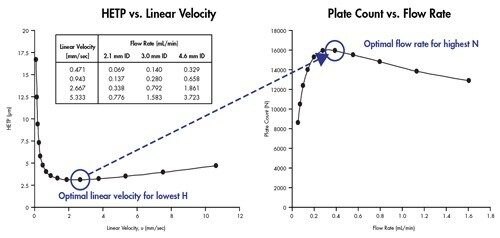
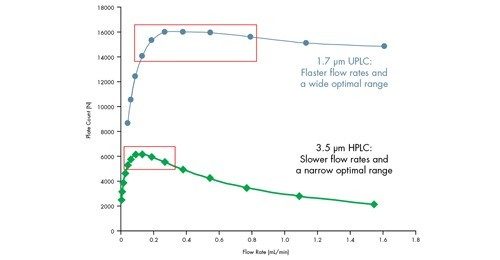
Eine Van-Deemter-Kurve wird durch Addition der Terme A, B und C gebildet [Abbildung 25]. Die Trennung bei der Lineargeschwindigkeit, bei der der niedrigste Punkt der Kurve auftritt, führt zur höchsten Effizienz und damit zur höchsten chromatographischen Auflösung.
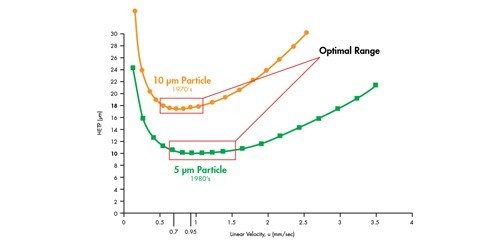
Wenn wir dies mit der in Abbildung 23 gezeigten Van-Deemter-Gleichung korrelieren, wird H um den Faktor 2 reduziert, wenn die Partikelgröße um die Hälfte reduziert wird. Daher ist es möglich, die Bandenverbreiterung innerhalb einer Säule durch Verwendung kleinerer Partikel zu reduzieren.
![Abbildung 14: Grundlegende Auflösungsgleichung [N] ist die Bodenzahl, [α] ist die Selektivität und [k] ist der Retentionsfaktor.](/content/dam/waters/de/figures/primers/uplc/primer_uplc_14.jpg.82.resize/img.jpg)
![Abbildung 16: Die Auflösung [Rs] ist direkt proportional zur Quadratwurzel der Effizienz [N].](/content/dam/waters/de/figures/primers/uplc/primer_uplc_16.jpg.82.resize/img.jpg)
![Abbildung 17: Bei konstanter Säulenlänge ist die Effizienz [N] umgekehrt proportional zur Partikelgröße [dp].](/content/dam/waters/de/figures/primers/uplc/primer_uplc_17.jpg.82.resize/img.jpg)
![Abbildung 18: Bei konstanter Säulenlänge ist die Flussrate [Fopt] umgekehrt proportional zur Partikelgröße [dp], was zu einer Verringerung der Analysezeit [T] proportional zur Verringerung der Partikelgröße führt.](/content/dam/waters/de/figures/primers/uplc/primer_uplc_18.jpg.82.resize/img.jpg)
![Abbildung 19: Schmalere Peakbreiten erzeugen eine höhere Effizienz und Peakhöhen. Die Effizienz [N] ist umgekehrt proportional zur Peakbreite [w] zum Quadrat. Außerdem nimmt mit abnehmender Peakbreite die Peakhöhe proportional zu.](/content/dam/waters/de/figures/primers/uplc/primer_uplc_19.jpg.82.resize/img.jpg)
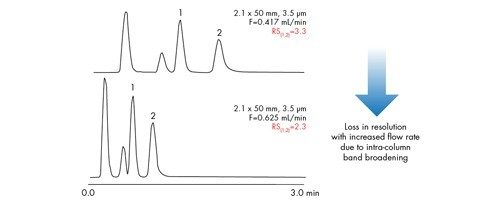
![Abbildung 20: Abgleich von Theorie und Realität. Trennungen wurden an zwei Säulen mit denselben Abmessungen [2,1 x 50 mm] durchgeführt. Bei beiden Trennungen wurden identische Chromatographiebedingungen verwendet, mit Ausnahme der Flussrate, die basierend auf der Partikelgröße skaliert wurde.](/content/dam/waters/de/figures/primers/uplc/primer_uplc_20.jpg.82.resize/img.jpg)
![Abbildung 21: Vereinfachte Gleichung zur Bestimmung von HETP. [L] ist die Säulenlänge, [N] ist die Bodenzahl und [HETP] ist die Höhe eines theoretischen Bodens.](/content/dam/waters/de/figures/primers/uplc/primer_uplc_21.jpg.82.165-6-331-51C.resize/img.jpg)
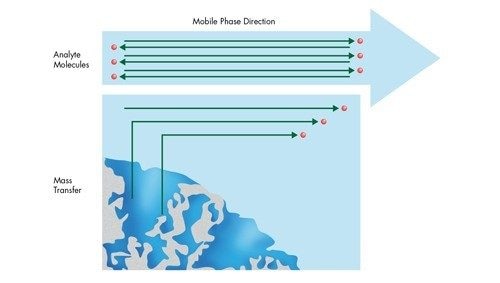
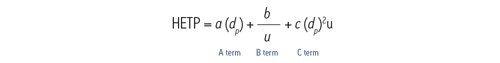

![Abbildung 29: Massentransfer [Diffusion] in eine und aus einer chromatographischen Pore](/content/dam/waters/de/figures/primers/uplc/primer_uplc_29.jpg.82.resize/img.jpg)
![Abbildung 30: Einfluss der linearen Geschwindigkeit auf Massentransfer und Analytbanden [gleiche Partikelgröße]](/content/dam/waters/de/figures/primers/uplc/primer_uplc_30.jpg.82.resize/img.jpg)
![Abbildung 31: Unterschiede des Massentransfers in Bezug auf die Partikelgröße [Darstellung einer 100Å-Pore]. Bei kleineren Partikeln werden schmalere Analytbanden gebildet.](/content/dam/waters/de/figures/primers/uplc/primer_uplc_31.jpg.82.resize/img.jpg)
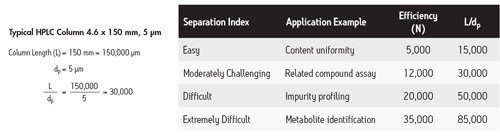
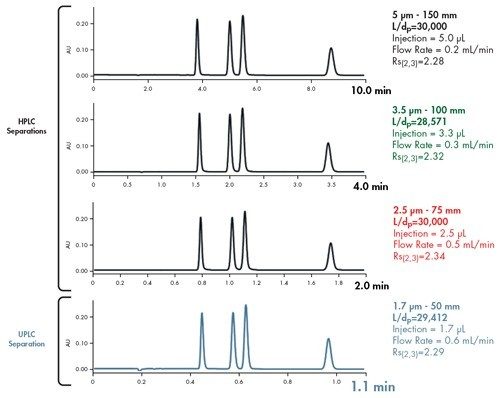
![Abbildung 39: Vergleich des L/dp-Verhältnisses als Funktion des Trennindex [einfach bis extrem schwierig]. Säulen mit demselben L/dp-Verhältnis erzeugen das gleiche Auflösungsvermögen.](/content/dam/waters/de/figures/primers/uplc/primer_uplc_39.jpg.82.resize/img.jpg)
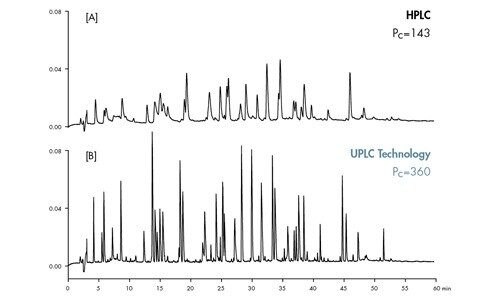
![Abbildung 41: Gleichung für die Peakkapazität [Pc], wobei [tg] die Gradientenzeit und [w] die durchschnittliche Peakbreite ist.](/content/dam/waters/de/figures/primers/uplc/primer_uplc_41.jpg.82.resize/img.jpg)
![Abbildung 42: Anwenden der Peakkapazitätsgleichung auf eine schnelle Trennung, wobei 0,37 Minuten die Gradientendauer und 0,01 Minuten die durchschnittliche Peakbreite sind, was zu einer Peakkapazität von 38 führt. Die Peakbreite wurde bei 13,4 % der Peakhöhe gemessen [4σ].](/content/dam/waters/de/figures/primers/uplc/primer_uplc_42.jpg.82.resize/img.jpg)