Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Accordance With EPA 1633 Part 1: Establishing and Assessing the Method
Abstract
US EPA Method 1633 has become the foundation method for analysis of PFAS in non-potable water matrices, soils, biosolids, and tissues in the United States. The method consists of sample preparation using weak anion exchange (WAX) solid phase extraction (SPE) with graphitized carbon black (GCB) clean up. This application note is the first in a series demonstrating a comprehensive solution for performing the 1633 methodology. The focus of this note is on establishing the LC-MS/MS method on an ACQUITY™ Premier BSM FTN LC System coupled to a Xevo™ TQ Absolute Tandem Quadrupole Mass Spectrometer and evaluating the method performance using waters_connect™ for Quantitation Software.
Benefits
- An 11-minute gradient method for analysis of 40 target analyte PFAS and 31 isotope labelled internal standards that meets the requirements of EPA 1633 for fast instrumental analysis time
- A stable and robust UPLC method using the ACQUITY Premier BSM FTN that surpasses retention time requirements of EPA 1633
- A sensitive and reliable MS/MS method using the Xevo TQ Absolute that fulfills requirements for initial and on-going calibration and ion ratio standards of EPA 1633
- Ease of data processing and reporting requirements with the use of waters_connect for Quantitation
Introduction
US EPA Method 1633 was first introduced in August 2021 to become the foundation method for analysis of PFAS in non-potable water matrices, soils, biosolids, and tissues.1 At the time of the writing of this document, Method 1633 is in the 4th Draft Phase with the final version expected to be released near the end of 2023. By the final release of EPA 1633 it will have been multi-lab validated for each type of sample matrix included in the method. The method covers 40 PFAS and utilizes isotope dilution calibration and quantitation. Required sample preparation differs slightly depending on sample type, but all sample types utilize solid phase extraction (SPE) on a weak anion exchange (WAX) cartridge in combination with graphitized carbon black (GCB) clean up. EPA 1633 was created to support sample analysis for the Clean Water Act (CWA) and Department of Defense (DoD) monitoring and remediation, but it covers such a wide range of matrices and compounds that its applicability can be widespread.
This is the first in a series of application notes addressing sample preparation, analysis, and method performance of EPA 1633 using a comprehensive workflow of Waters technologies. This application note will focus on establishing the LC-MS/MS method on an ACQUITY Premier BSM FTN LC System coupled to a Xevo TQ Absolute Mass Spectrometer and evaluating the method performance using waters_connect for Quantitation Software. Future application notes will cover method recoveries and authentic sample analysis.
Experimental
Sample Preparation
Details of the sample preparation are thoroughly discussed in the 2nd and 3rd application notes in this series. In brief, the sample preparation outlined in 1633 was followed using Waters™ Oasis™ WAX for PFAS in combination with GCB cleanup. One notable exception was for aqueous samples, a sample volume of 250 mL was used instead of 500 mL. This resulted in a 50x concentration factor for samples rather than 100x. The sensitivity of the Xevo TQ Absolute MS allowed for the reduction of sample volume. The following data presented will demonstrate that the 250 mL sample size produces equivalent results to those obtained in EPA 1633 using a 500 mL sample size. The benefits of a smaller sample volume include not only faster sample loading during the sample preparation process, but also reduced shipping costs and sample storage requirements when using the smaller 250 mL sample bottles.
All standards used (target analytes, extracted internal standards, and non-extracted internal standards) were mixes from Wellington Laboratories specifically developed for EPA method 1633.
Samples discussed in this application note include ground water and surface water that were collected locally, and influent and effluent wastewater that were kindly provided by a municipal wastewater treatment facility in the Midwest United States.
Data Review
Data processing and review was performed using the MS Quan application in waters_connect for Quantitation. The following data quality guidelines outlined in EPA 1633 draft 4 will be highlighted to demonstrate method capability using the Waters solutions:
1. The mid calibration point was used as the reference point (designated as Quan Reference in waters_connect) for retention time and ion ratios.
2. Retention times must be within 0.4 min of reference point.
3. Target analytes must elute within 0.1 min of Extracted Internal Standard (EIS).
4. Bile salts (TDCA, TCDCA, TUDCA) must be resolved 1 min from PFOS retention time window.
5. A minimum of 6 calibration points is required.
6. The lowest calibration point must have a S:N of 3 for quantitation and confirmation ions OR S:N of 10 if only one quantitation ion is available.
7. When using average RF calibration, the %RSD must be ≤20% to establish linearity.
8. Calibration Verification (CV) injections must be within 70–130% of the expected concentration.
9. Ion Ratios (where applicable) must be within 50–150% of mid-point calibration reference point.
10. The Minimum Level of Quantitation (ML) established is the lowest level at which the LC-MS/MS gives a recognizable signal and acceptable calibration point for the analyte. This is the lowest concentration an analyte can be measured at for the method.
11. Linear and branched isomers are reported as a single summed result.
LC Conditions
|
LC system: |
ACQUITY Premier BSM with FTN |
|
Vials: |
700 µL Polypropylene Screw Cap Vials (p/n: 186005219) |
|
Analytical column: |
ACQUITY Premier BEH™ C18 2.1 x 50 mm, 1.7 µm (p/n: 186009452) |
|
Isolator column: |
Atlantis Premier BEH C18 AX 2.1 x 50 mm, 5.0 µm (p/n: 186009407) |
|
Column temperature: |
35 °C |
|
PFAS kit: |
PFAS Install Kit with OASIS WAX 150 mg (p/n: 176004548) |
|
Sample temperature: |
10 °C |
|
Injection volume: |
2 µL |
|
Flow rate: |
0.3 mL/min |
|
Mobile phase A: |
2 mM ammonium acetate in water |
|
Mobile phase B: |
2 mM ammonium acetate in acetonitrile |
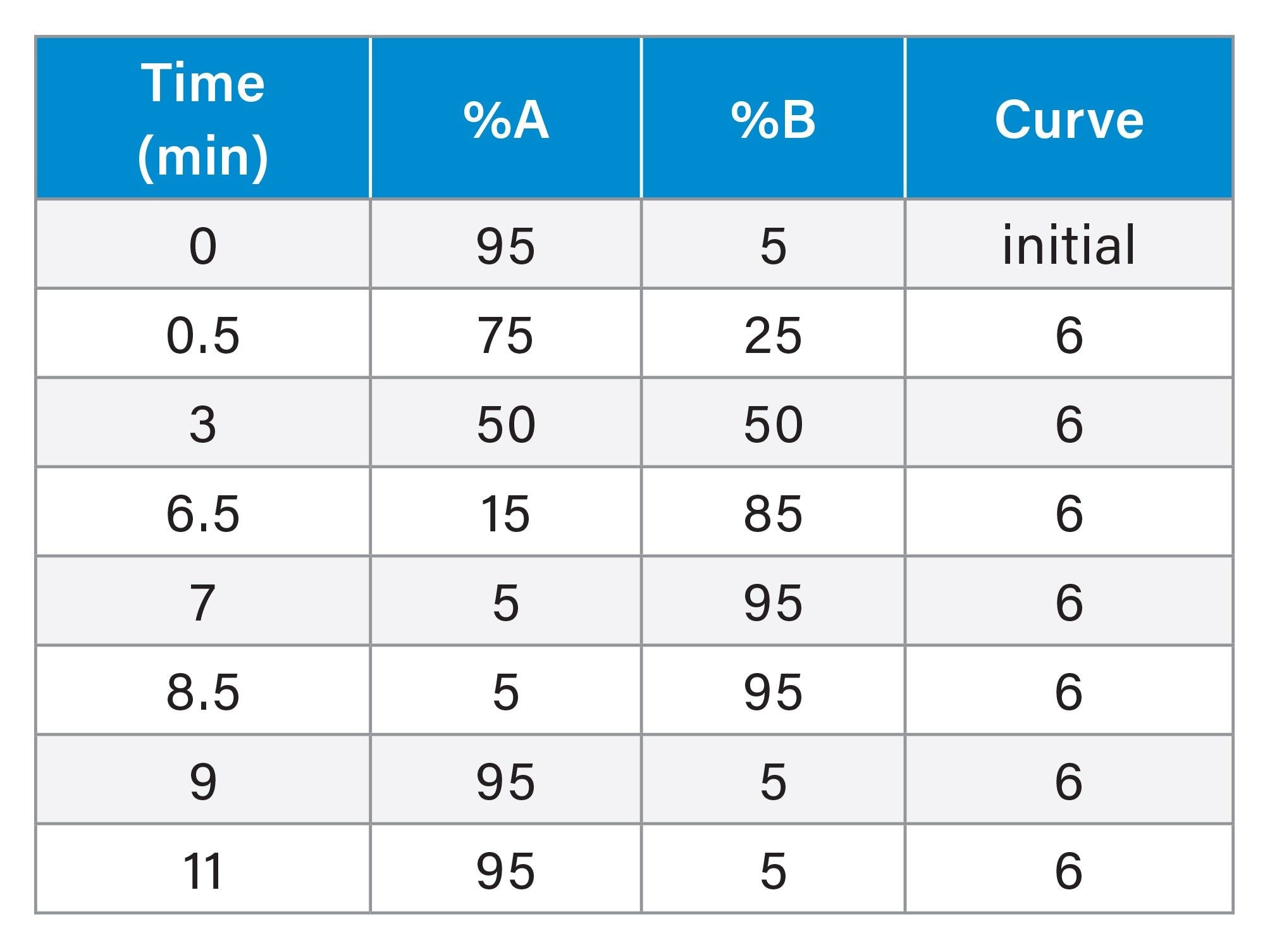
Gradient Table
MS Conditions
|
MS system: |
Xevo TQ Absolute |
|
Ionization mode: |
ESI- |
|
Capillary voltage: |
0.5 kV |
|
Source temperature: |
100 °C |
|
Desolvation temperature: |
350 °C |
|
Desolvation flow: |
900 L/hr |
|
Cone flow: |
150 L/hr |
|
MRM method: |
See Appendix for Full MRM Method details |
Data Management
|
Software: |
waters_connect for Quantitation |
Results and Discussion
LC Gradient Optimization
Cholic acids, such as taurodeoxycholic acid (TDCA), taurochenodeoxycholic acid (TCDCA), and tauroursodeoxycholic acid (TUDCA), can cause interference with PFOS in the mass spectrometer due to the similarity in parent and fragment masses. These interferents are bile salts produced to aid the digestion process which means that they can be present in tissue and wastewater samples. Therefore, one of the requirements of the LC method for EPA 1633 is to not only monitor cholic acids, but also to ensure there is a one-minute retention time difference between the cholic acids and PFOS. Previous PFAS methods published have used methanol as the organic mobile phase.2,3 When the methanol method was tested for use with EPA 1633 samples, PFOS eluted in the middle of the three cholic acids (Figure 1). This would not satisfy the requirements of 1633, so acetonitrile was tested as the organic mobile phase. As can be seen in Figure 1, using acetonitrile, the cholic acids elute much earlier than any of the PFOS isomers, providing greater than a one-minute separation. It should be noted that a small reduction in response was noticed when switching from methanol to acetonitrile, but not enough to affect the analysis.
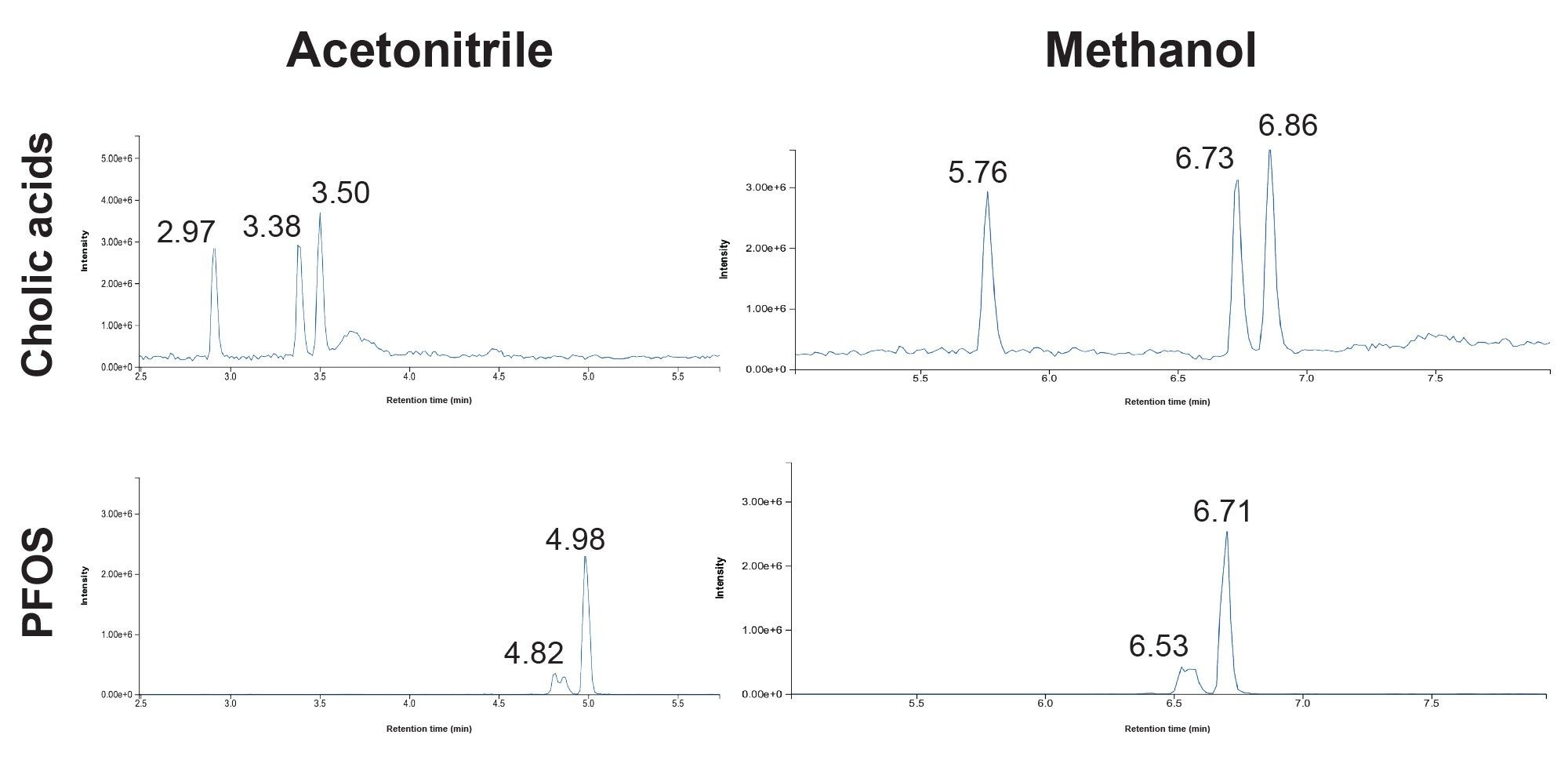 Figure 1. Comparison of the resolution of cholic acids from PFOS isomers using acetonitrile (left) and methanol (right) as the organic mobile phase.
Figure 1. Comparison of the resolution of cholic acids from PFOS isomers using acetonitrile (left) and methanol (right) as the organic mobile phase.
In addition to changing the organic mobile phase, the LC gradient method was scaled down from previous methods using a 100 mm column to a 50 mm column to decrease instrumental analysis time.
Calibration Performance and Ongoing Verification
EPA 1633 was developed, and single-laboratory validated using the Xevo TQ-S micro Mass Spectrometer, a tandem quadrupole mass spec with fit-for-purpose sensitivity. The work being presented here was performed using the highest sensitivity mass spec, the Xevo TQ Absolute Mass Spectrometer. The sensitivity level of the Xevo TQ Absolute MS allows for more method flexibility. Therefore, the calibration range used in this work was 20x lower than that used in the method and multi-lab validation study. Additionally, the minimum level of quantitation obtained on the Xevo TQ Absolute is approximately 20x lower as well. The calibration range (represented both as in vial and in sample concentrations) used for this work can be seen in Table 1. Also shown in Table 1 is the signal:noise (S:N) values of each compound at the minimum level of quantitation. All compounds had a S:N ≥3 at this level.
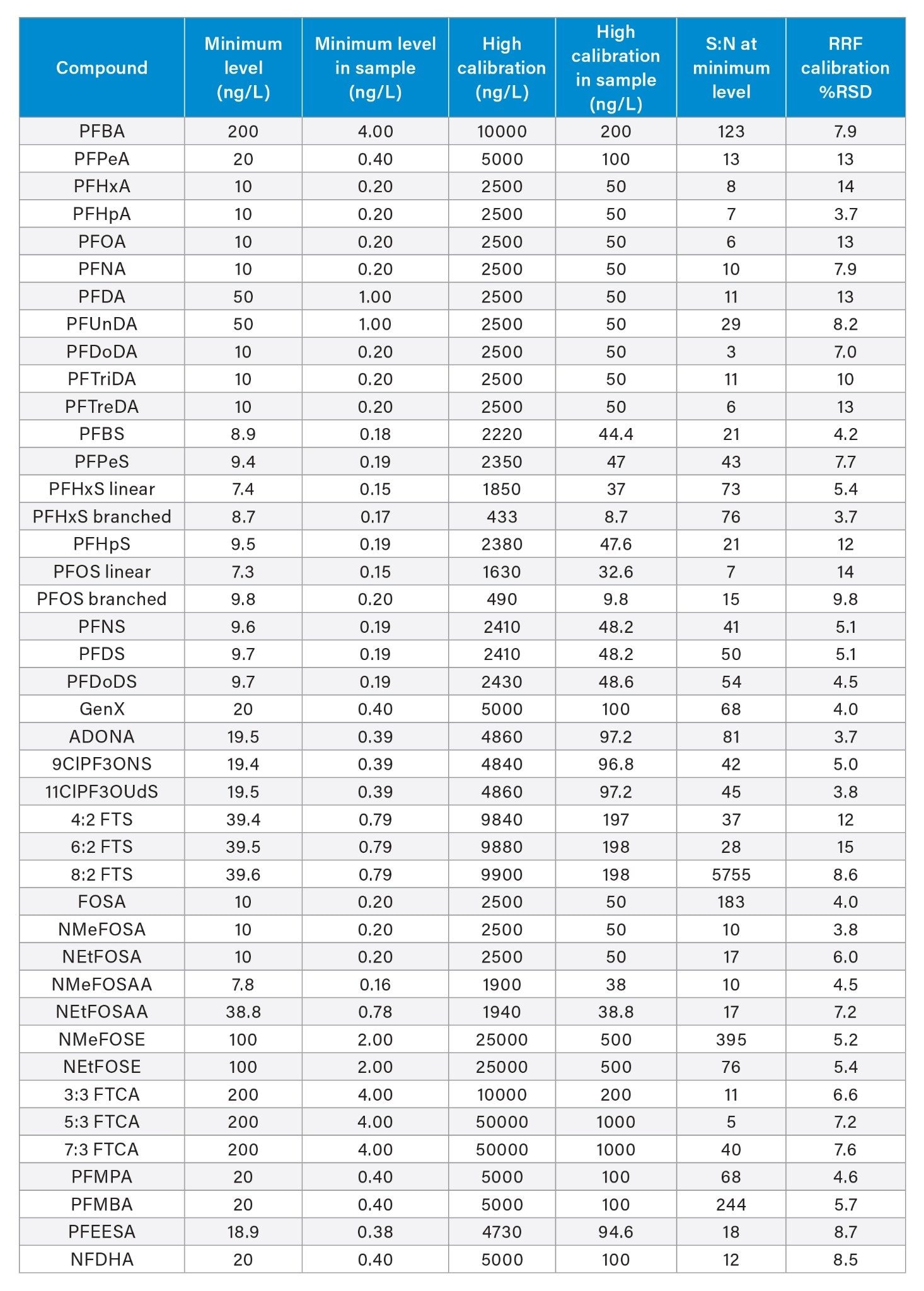 Table 1. Calibration curve data used for EPA 1633 evaluation on the Xevo TQ Absolute MS.
Table 1. Calibration curve data used for EPA 1633 evaluation on the Xevo TQ Absolute MS.
Calibration and quantification were performed using isotope dilution as instructed by the method. The calibration curve was plotted using Average Response Factor (RF). Using this type of curve, linearity is established by demonstrating %RSD of the Relative Response Factor (RRF) is ≤20% over the entire curve. The %RSD of RRF values are presented in Table 1, with all compounds having a %RSD <15%, demonstrating linearity of the calibration curves for all PFAS in the method.
Since a calibration curve is not required for every batch of samples, a Calibration Verification (CV) injection before each batch, after every ten samples of a batch, and at the end of a batch verifies the instrument stability during sample analysis. The CV is a calibration standard at the mid-level of the calibration curve. All CVs must quantify within 70–130% of the expected concentration for the calibration curve to be acceptable for use with each batch of data. To demonstrate instrument response stability, the % Deviation of the CV’s injected over a period of eight days run during various batches of ground water, surface water, wastewater, and tissue samples are plotted in Figure 2. To fall within the requirements of 1633, the % Deviation should fall within ±30% in this figure. All compounds, with the exception of 7:3 FTCA in two injections, fit the 30% deviation criteria. 7:3 FTCA had two injections where the deviation was >30%. Excluding 7:3 FTCA, the mean deviation of the CV values was 3.75%. This indicates that the LC-MS/MS system and methods are stable over a long period of time.
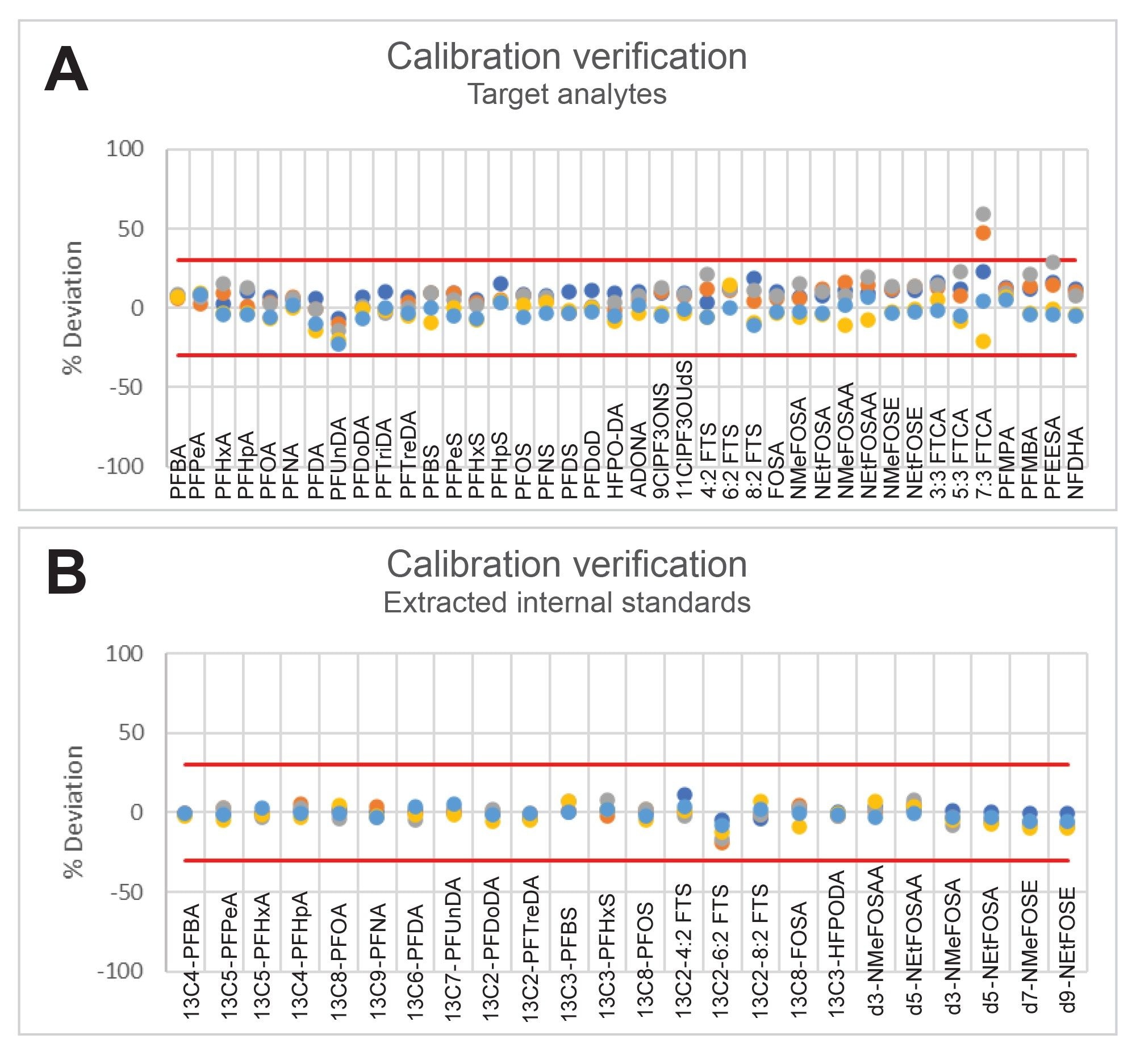 Figure 2. % Deviation of each injection of a CV injected during batches run over a period of 8 days for target analytes (A) and extracted internal standards (B). The red lines indicate the ±30% acceptance range.
Figure 2. % Deviation of each injection of a CV injected during batches run over a period of 8 days for target analytes (A) and extracted internal standards (B). The red lines indicate the ±30% acceptance range.
Method Performance During Sample Analysis
Retention time stability is another parameter requirement of EPA 1633, with an allowable RT window within 0.4 min of the expected retention time (either from initial calibration or CV). Figure 3 demonstrates the stability of the RTs of the acquired data over a range of 53 injections run during various batches of aqueous sample types over an 8-day time range. With the exception of FOSA (C8 sulfonamide), the maximum RT deviation of all compounds in all injections is 2.7% (0.02 min). The RT stability is demonstrated in this figure by the symmetry of the lines, with each line matching around the circle graph from its beginning (injection 1) to its end (injection 53). There is a noticeable shift in retention time of FOSA and 13C8-FOSA during injections 18 through 33, but the RT does stabilize back to the expected RT for subsequent injections. The 0.18 min RT shift for this compound is still well within the allowable 0.4 min range. Additionally, the RTs for FOSA and 13C8-FOSA match during this shift.
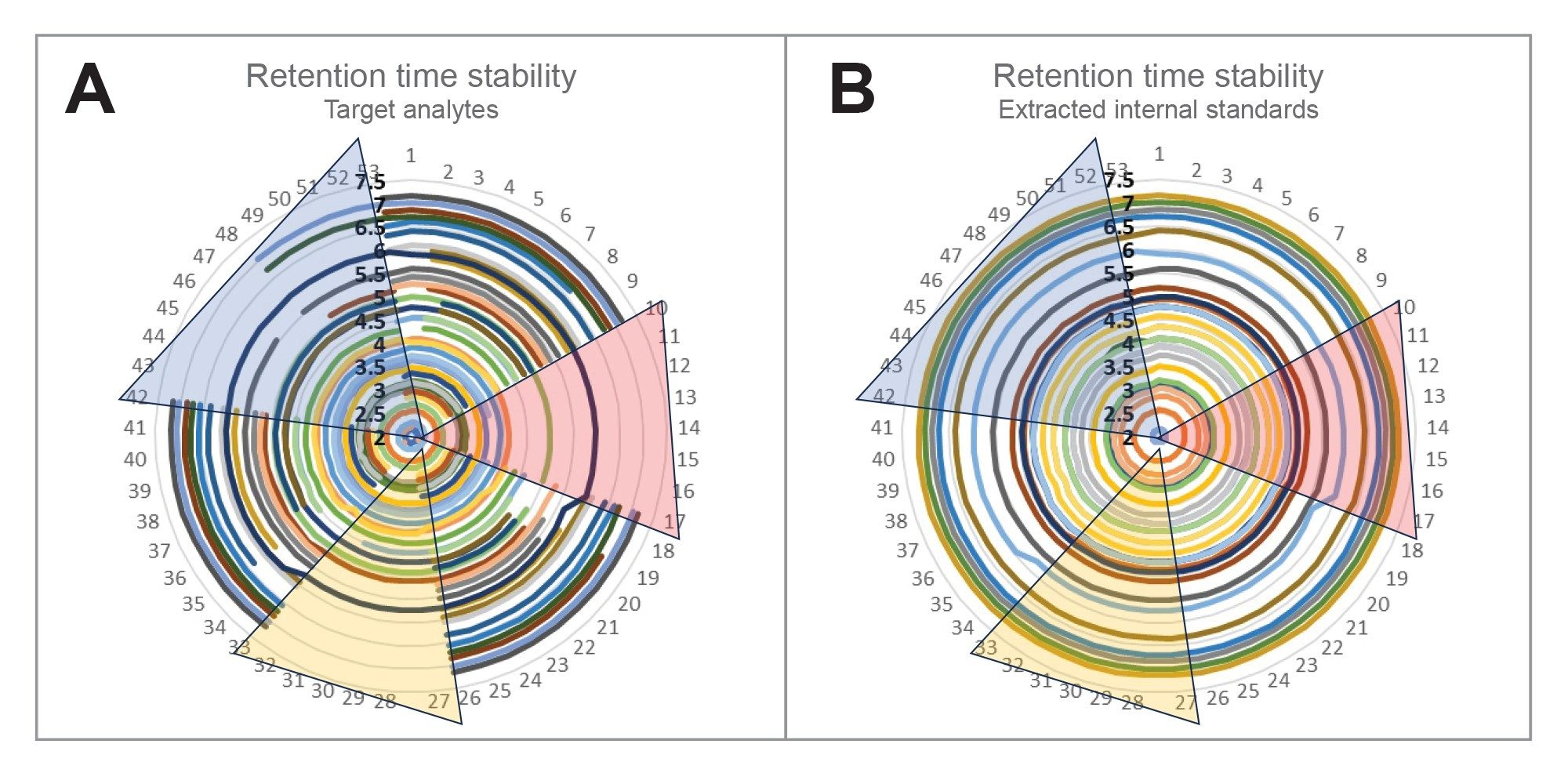 Figure 3. Retention time stability of target analytes (A) and extracted internal standards (B) over 53 injections of samples, standards, and quality control samples. The highlighted regions indicate regions of the graph that represent sample injections where red represents ground water, yellow represents surface water, and blue represents both influent and effluent wastewater.
Figure 3. Retention time stability of target analytes (A) and extracted internal standards (B) over 53 injections of samples, standards, and quality control samples. The highlighted regions indicate regions of the graph that represent sample injections where red represents ground water, yellow represents surface water, and blue represents both influent and effluent wastewater.Finally, ion ratios were measured over the 8 days of ground water, surface water, waste water, and tissue injections to measure the stability of ion ratios. EPA 1633 requires the ion ratios to be within 50–150% of the reference (mid-point or CV). The ion ratio deviation measured in Figure 4 is in comparison to the initial mid-point calibration. All ion ratios for all compounds were within the ±50% deviation range, with a mean deviation of 3.28%.
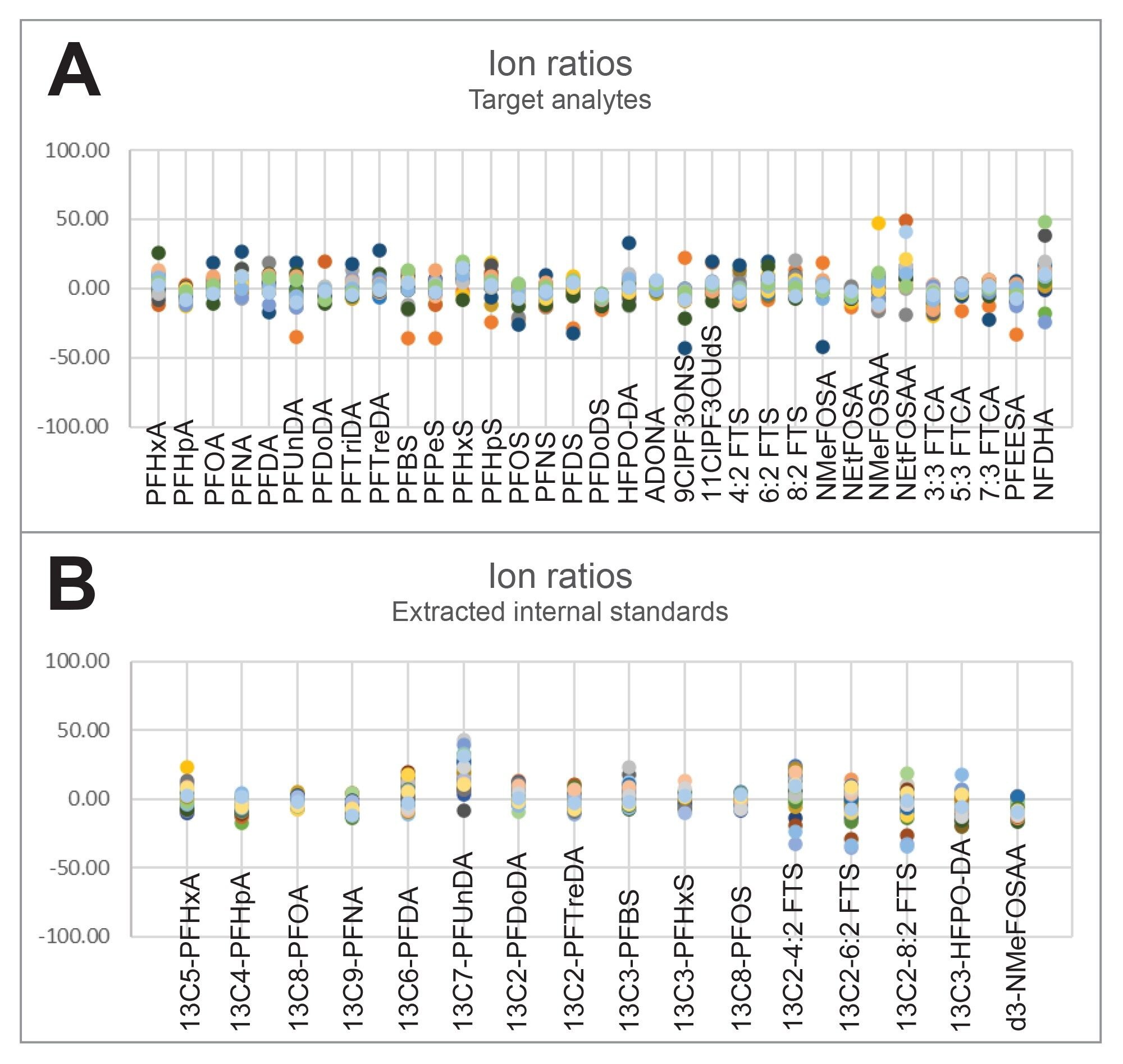 Figure 4. % Deviation of ion ratios calculated for each analyte having two MRM transitions for target analytes (A) and extracted internal standards (B). Data plotted represents injections of samples and standards during batches run over a period of eight days.
Figure 4. % Deviation of ion ratios calculated for each analyte having two MRM transitions for target analytes (A) and extracted internal standards (B). Data plotted represents injections of samples and standards during batches run over a period of eight days.
Data Processing With Isomers
An additional requirement of EPA 1633, which is a common requirement of most PFAS methods, is to sum branched and linear isomers together in the final reported results. Using the MS Quan app in waters_connect for Quantitation Software, totaling isomers is a simple and automated process. Figure 5 demonstrates the ability to independently process and quantify the isomers as well as the reporting of the total isomer concentration using PFHxS as an example.
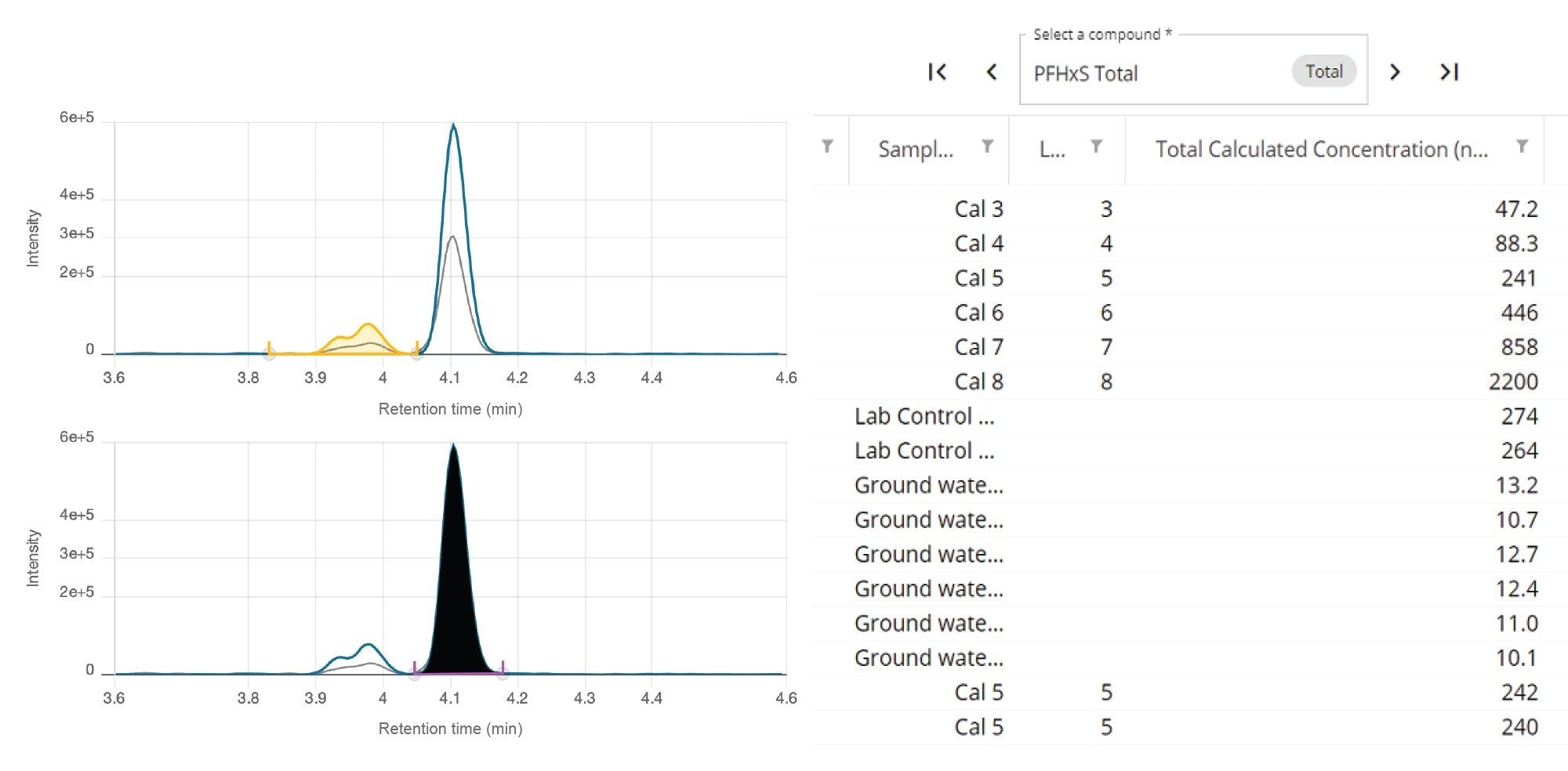 Figure 5. Individual isomer peak integration and reporting of total isomer concentration for PFHxS from MS Quan.
Figure 5. Individual isomer peak integration and reporting of total isomer concentration for PFHxS from MS Quan.
Conclusion
All data quality requirements from the EPA 1633 method were easily achieved using the analysis method presented on the ACQUITY Premier BSM FTN LC System coupled to Xevo TQ Absolute Mass Spectrometer. An 11-minute LC gradient on a 50 mm ACQUITY BEH C18 Column using acetonitrile as the organic mobile phase was able to produce the required one-minute resolution between potential cholic acid interferences and PFOS. The Xevo TQ Absolute allows for a considerably lower detection and calibration range than presented in EPA 1633. This presents the opportunity to detect lower concentrations of PFAS in samples and opens up the possibility to reduce sample size required, therefore speeding up sample preparation. Calibration was shown to be stable within the calibration verification requirements of ±30% response. Retention times were stable, within 2.7% deviation from the reference injection. Ion ratios were all well within the ±50% deviation as well. The data presented demonstrates that the LC-MS/MS system easily fulfills all requirements for use in sample analysis for EPA 1633.
References
- US Environmental Protection Agency. Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Aqueous, Solid, Biosolids, and Tissue Samples by LC-MS/MS, Draft 4. July 2023.
- K Organtini, K Rosnack, D Stevens, E Ross. Analysis of Legacy and Emerging Perfluorinated Alkyl Substances (PFAS) in Environmental Waters Samples Using Solid Phase Extraction (SPE) and LC-MS/MS. Waters Application Note. 720006471. Revised November 2022.
- K Organtini, H Foddy, N Dreolin, S Adams, K Rosnack, P Hancock. Ultra-trace Detection of Per- and Polyfluoroalkyl Substances (PFAS) in Drinking Water to meet New US EPA Interim Health Advisory Levels. Waters Application Note. 720007855. February 2023.
Appendix
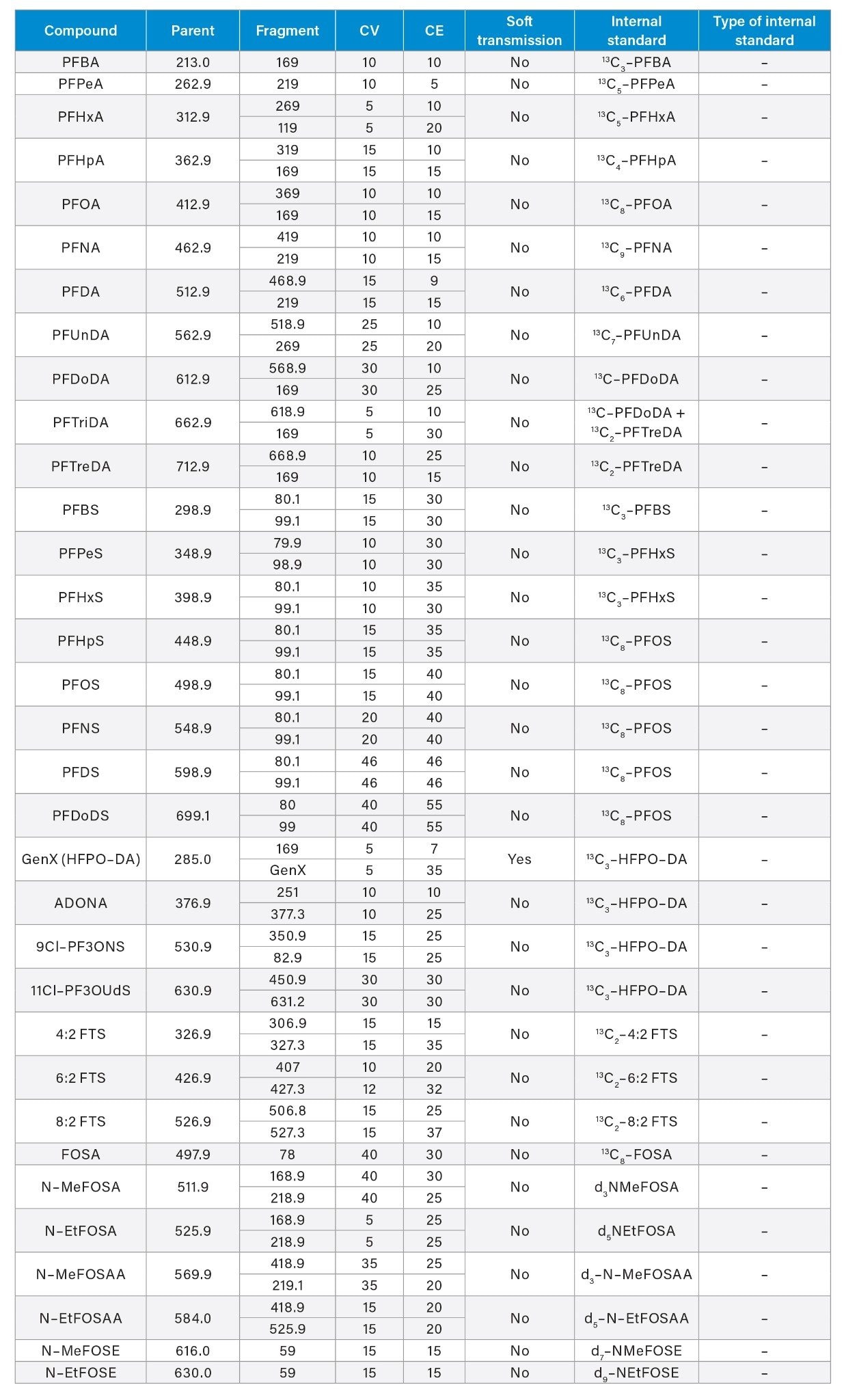
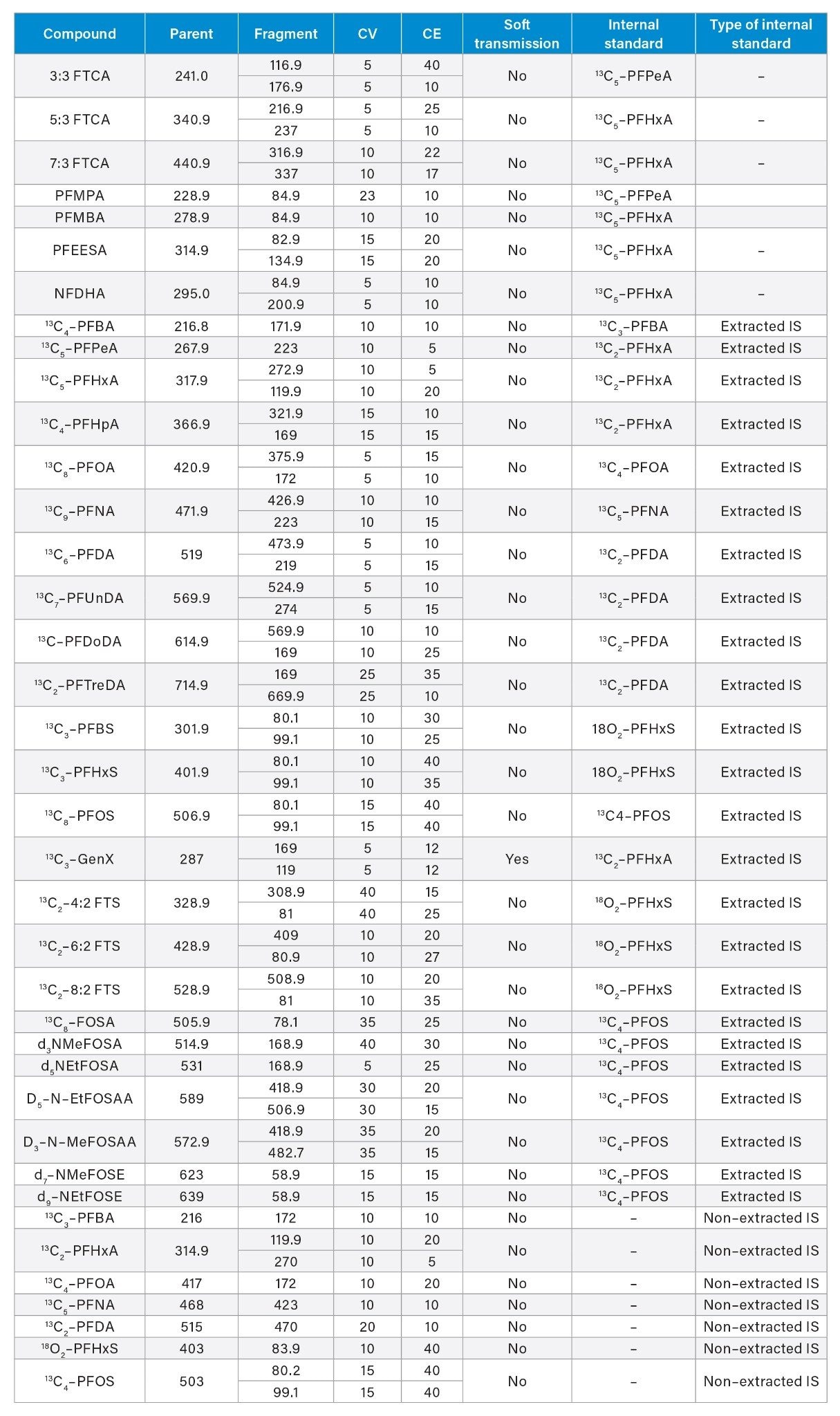 Appendix Table 1. MS Method conditions for PFAS included in analysis for EPA 1633.
Appendix Table 1. MS Method conditions for PFAS included in analysis for EPA 1633.
720008117, October 2023