Correct protein conformation is essential for proper biological function of protein therapeutics. Changes in protein conformation are a major concern in the biopharmaceutical industry and conformational characterization is a difficult task. Many analytical tools such as circular dichroism (CD), differential scanning calorimetry (DSC), and analytical ultracentrifugation (AUC) are used to study the higher-order structure of proteins. These methods sample global conformation and cannot determine where conformational changes occur. Nuclear magnetic resonance (NMR) and X-ray crystallography determine protein structure with high spatial resolution, but both technologies require substantial amounts of sample and are not routinely applied to biopharmaceutical products due to the significant time and effort required.
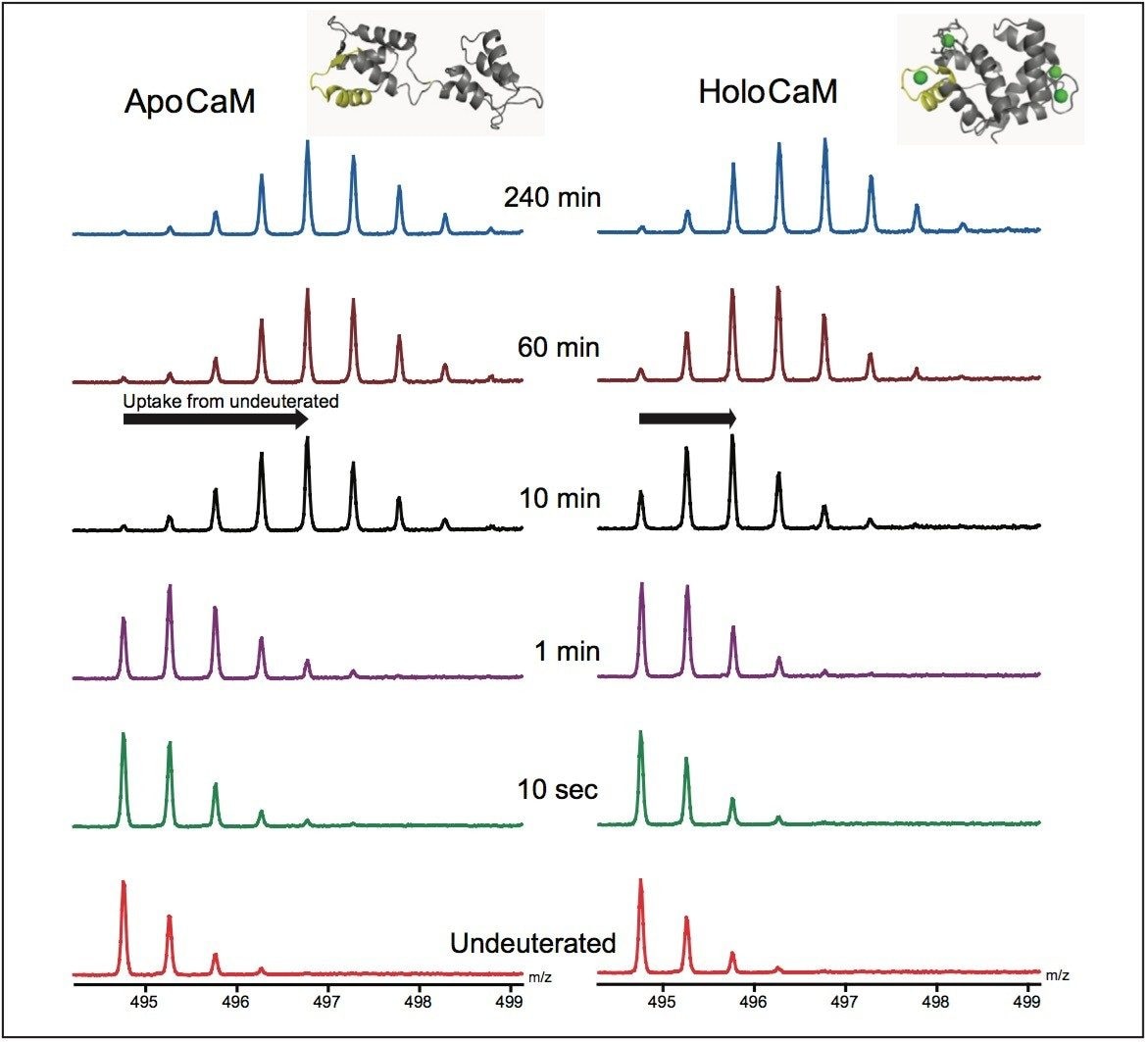
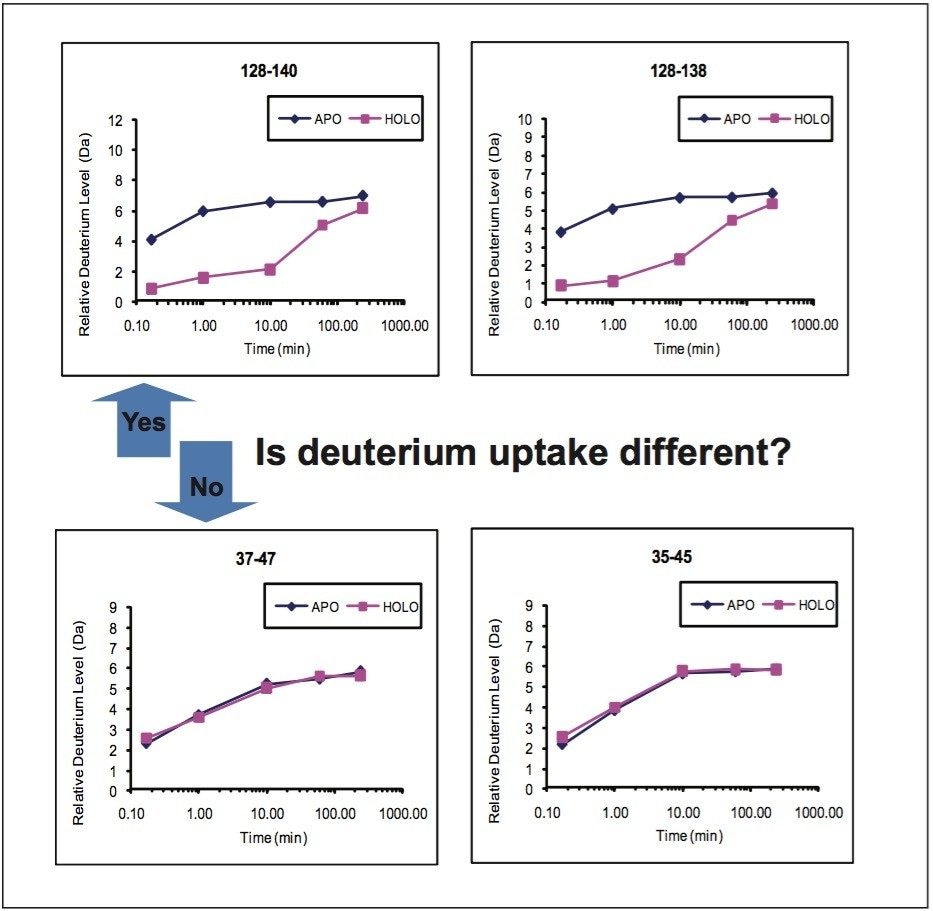
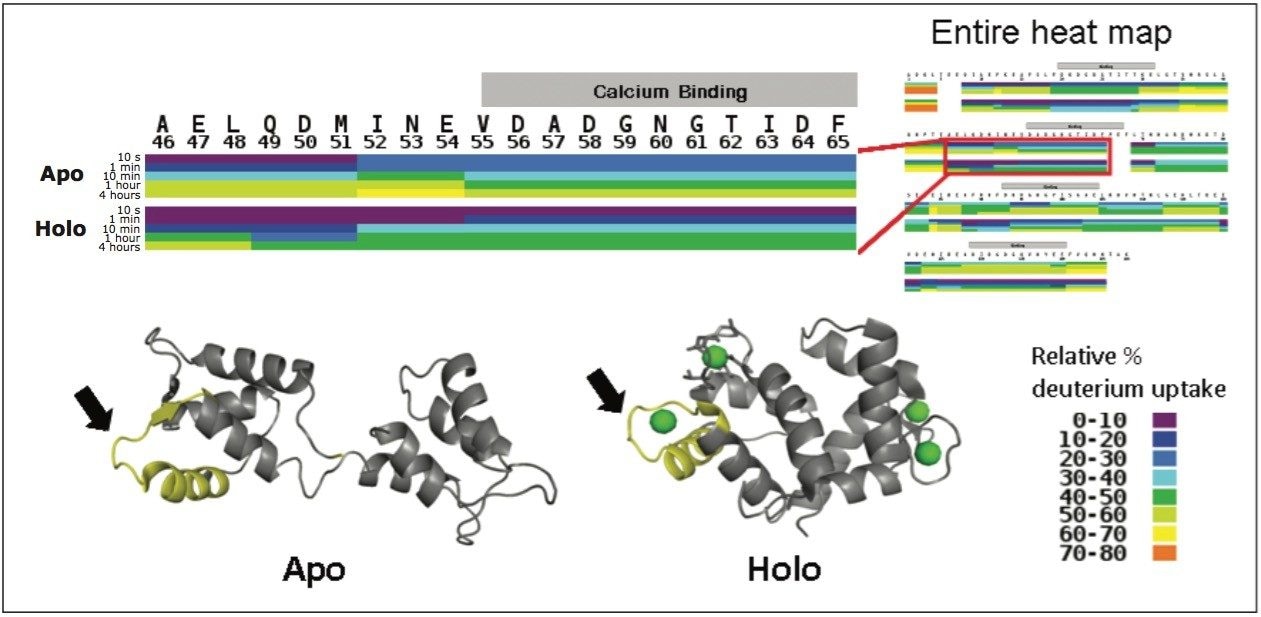
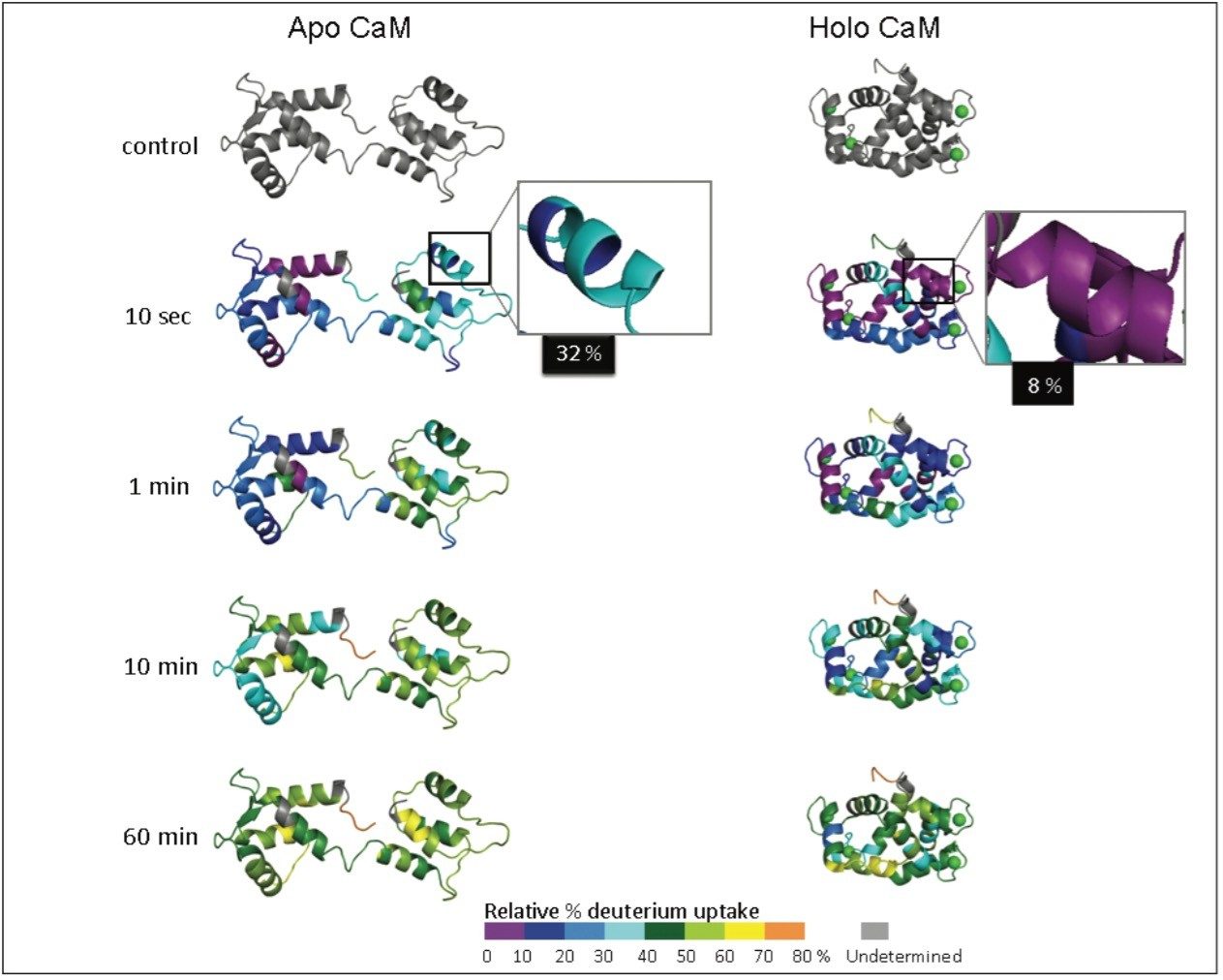
Hydrogen deuterium exchange (HDX or HX MS) mass spectrometry has proven to be a useful analytical method for the study of protein dynamics and changes to protein conformation.1 Successful HDX studies require an LC-MS system that can perform rapid chromatographic separations at 0 °C and make accurate mass measurements of small quantities of deuterium labeled proteins and peptides.2
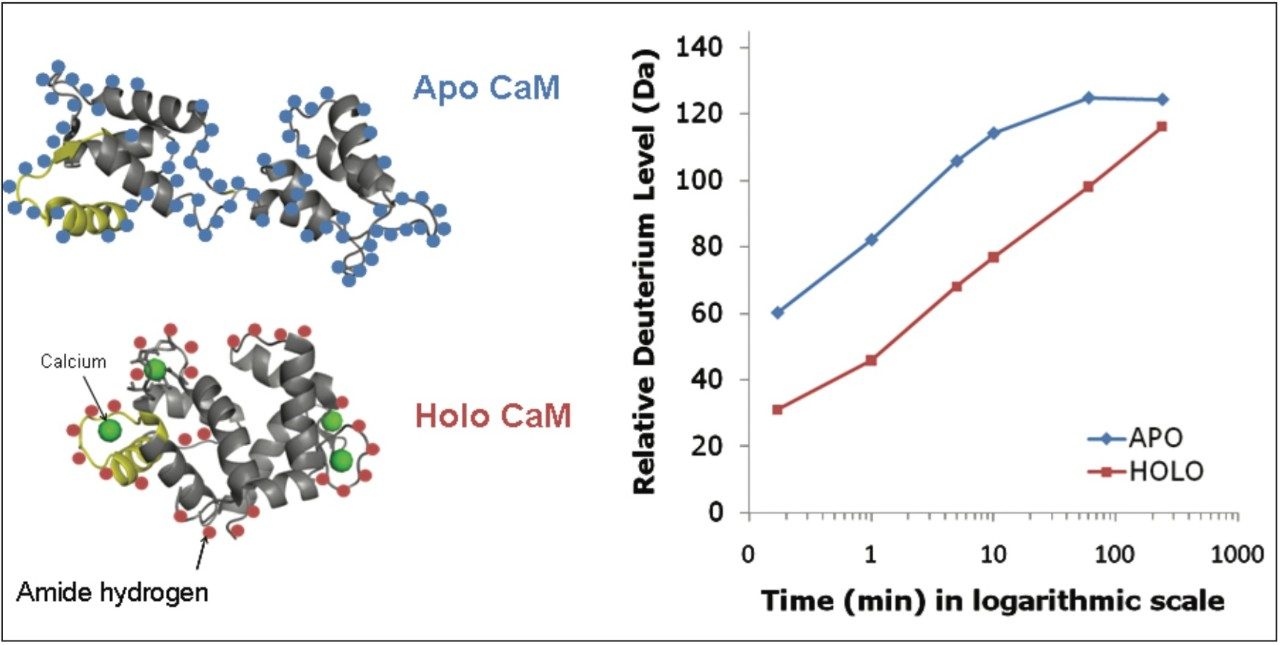
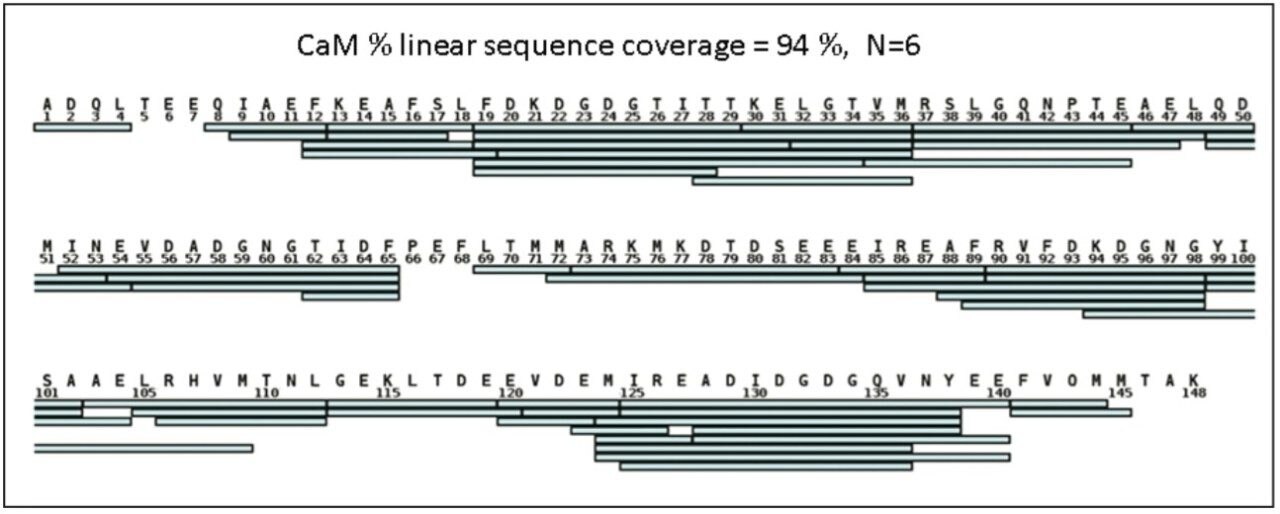
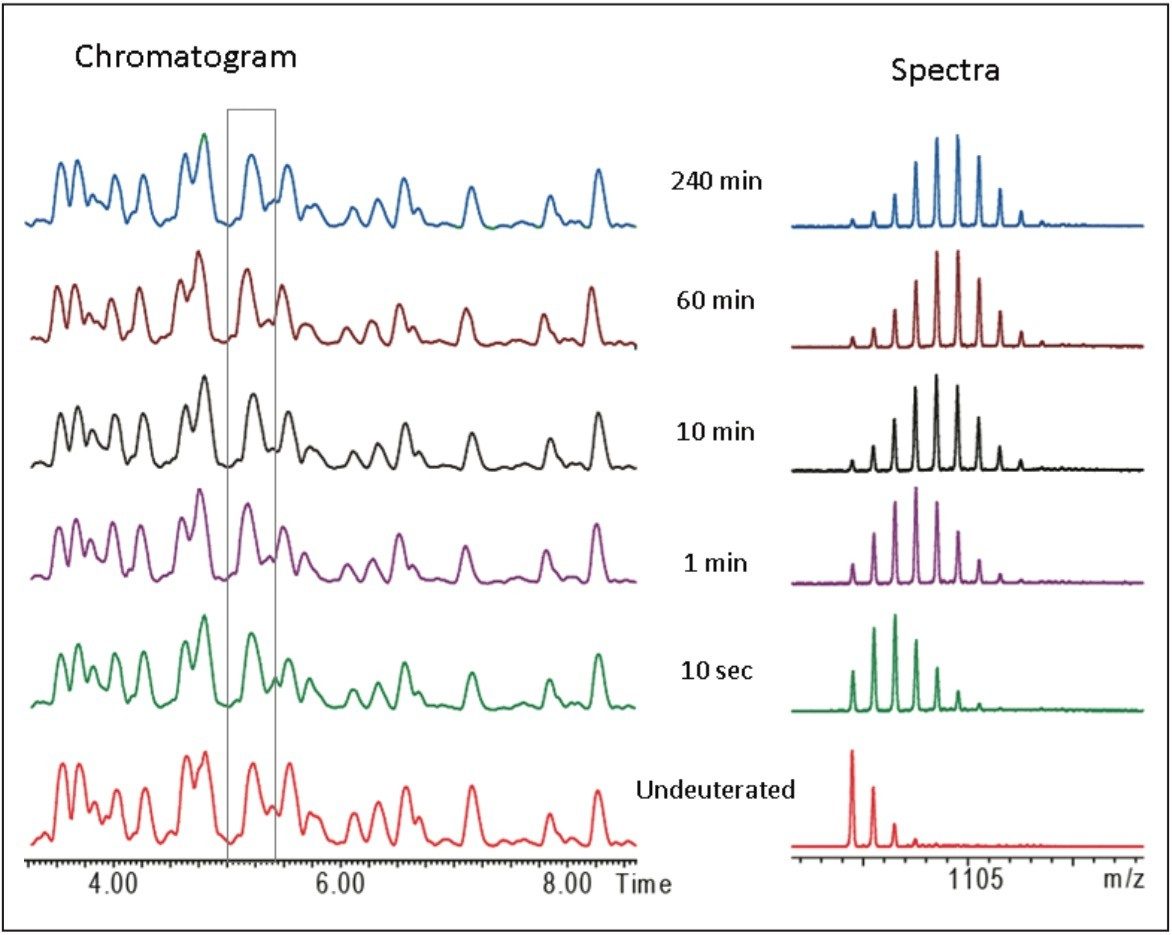
When an HDX experiment is performed with UPLC separation technology and high-resolution mass spectrometry, subtle conformational changes can be revealed and the locations of these changes are determined at the peptide level. Such innovative technology with a commercially available system solution makes conformational analyses a more practical method. Waters HDX Technology has been adopted as an effective analytical tool for biotech researchers who are studying higher-order structure of protein drugs. Characterizing higher-order structure is one of the key factors in drug discovery and early development phases and adopting this technology will offer a distinct advantage in understanding the safety and efficacy of biopharmaceuticals.
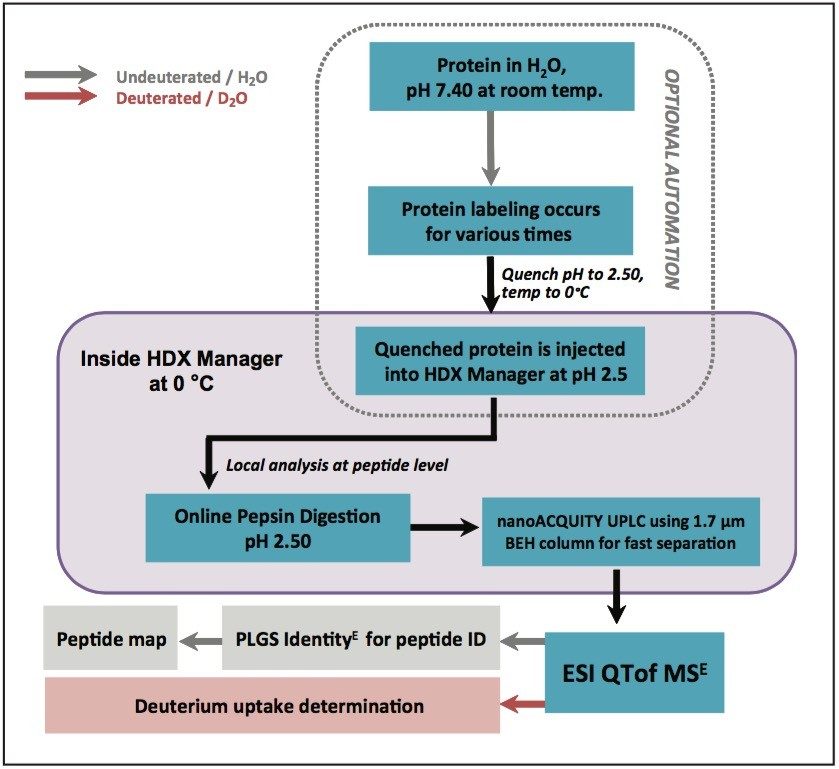
In this application note, we introduce the nanoACQUITY UPLC System with HDX Technology (Figure 1). This system consists of a nanoACQUITY UPLC Binary Solvent Manager (BSM), Auxiliary Solvent Manger (ASM), and HDX Manager. The HDX Manager is capable of rapid online protein digestion, desalting, and highly resolving chromatographic separations at 0 °C. Operating at 0 °C is required to reduce the deuterium loss during analyses. With this system, subtle differences in deuterium uptake can be adequately determined with very reproducible HDX conditions. A Xevo QTof MS, utilizing MSE with ProteinLynx Global SERVER (PLGS) Software, provides an ideal tool for accurate mass analysis and reliable peptic peptide assignment.