Acrylamide is a highly polar, water soluble compound and is formed during food production by high temperature (+120 °C) cooking with the main chemical reaction that causes this known as the Maillard Reaction.1 The levels in food are of concern due to the toxicological properties which include neurotoxicity, genotoxicity, carcinogenicity, and reproductive toxicity.1 As acrylamide is present in a wide range of everyday foodstuffs, an accurate and robust method that can detect and quantify low levels of acrylamide is vital. EU regulation 2017/2158 establishes mitigation measures and benchmark levels for reducing the presence of acrylamide in food.2
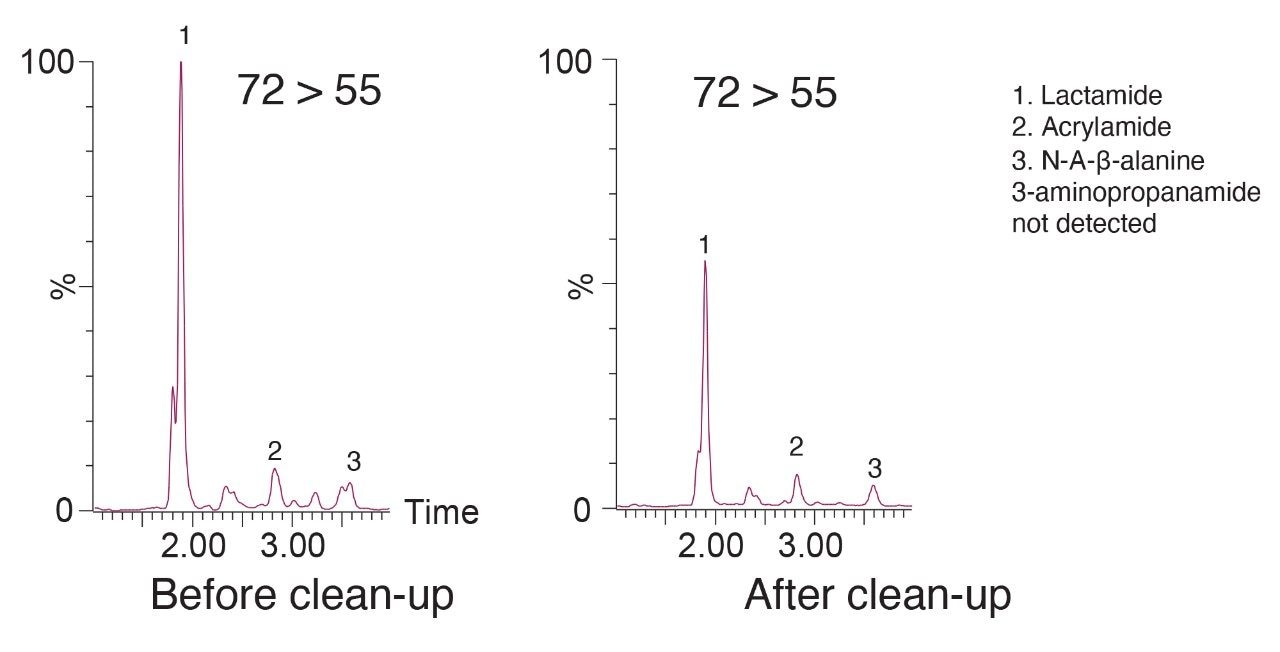
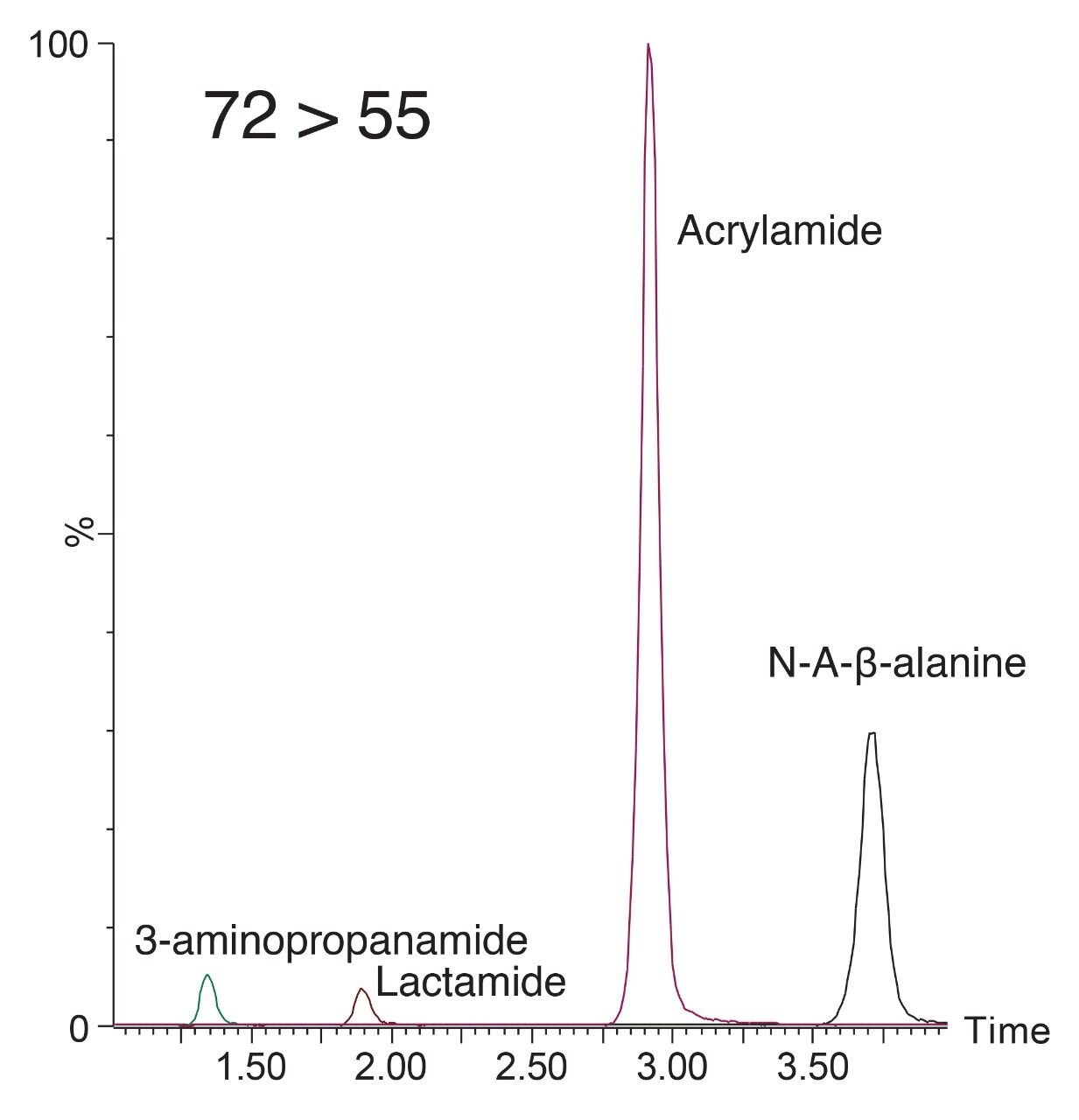
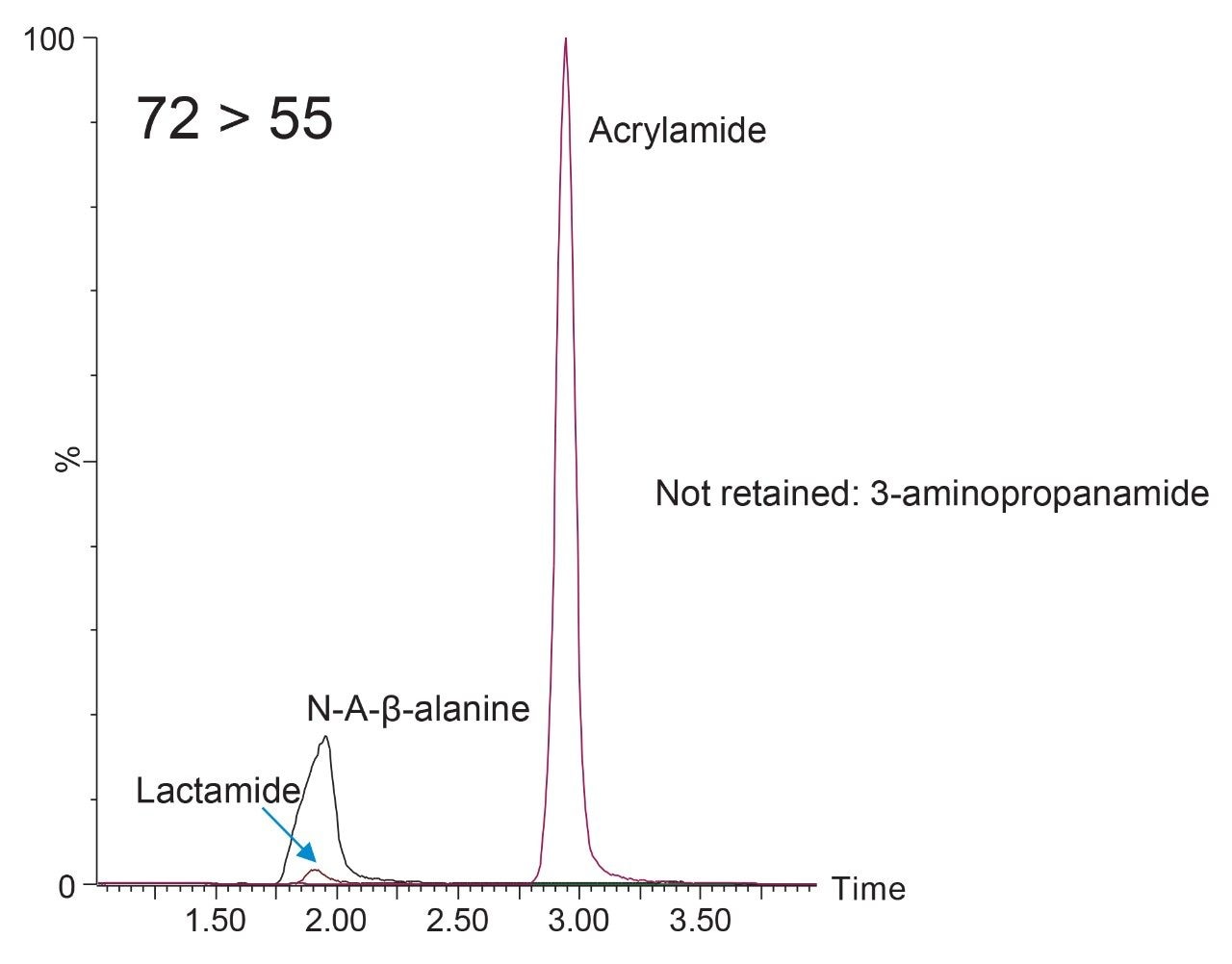
Our latest solution for determination of acrylamide in complex food matrices such as potato chips and coffee, utilizes a modified QuEChERS extraction procedure with two clean-up options, dispersive SPE or Oasis MCX pass through SPE for enhanced clean-up, prior to LC-MS/MS using an ACQUITY UPLC HSS C18 SB Column.3
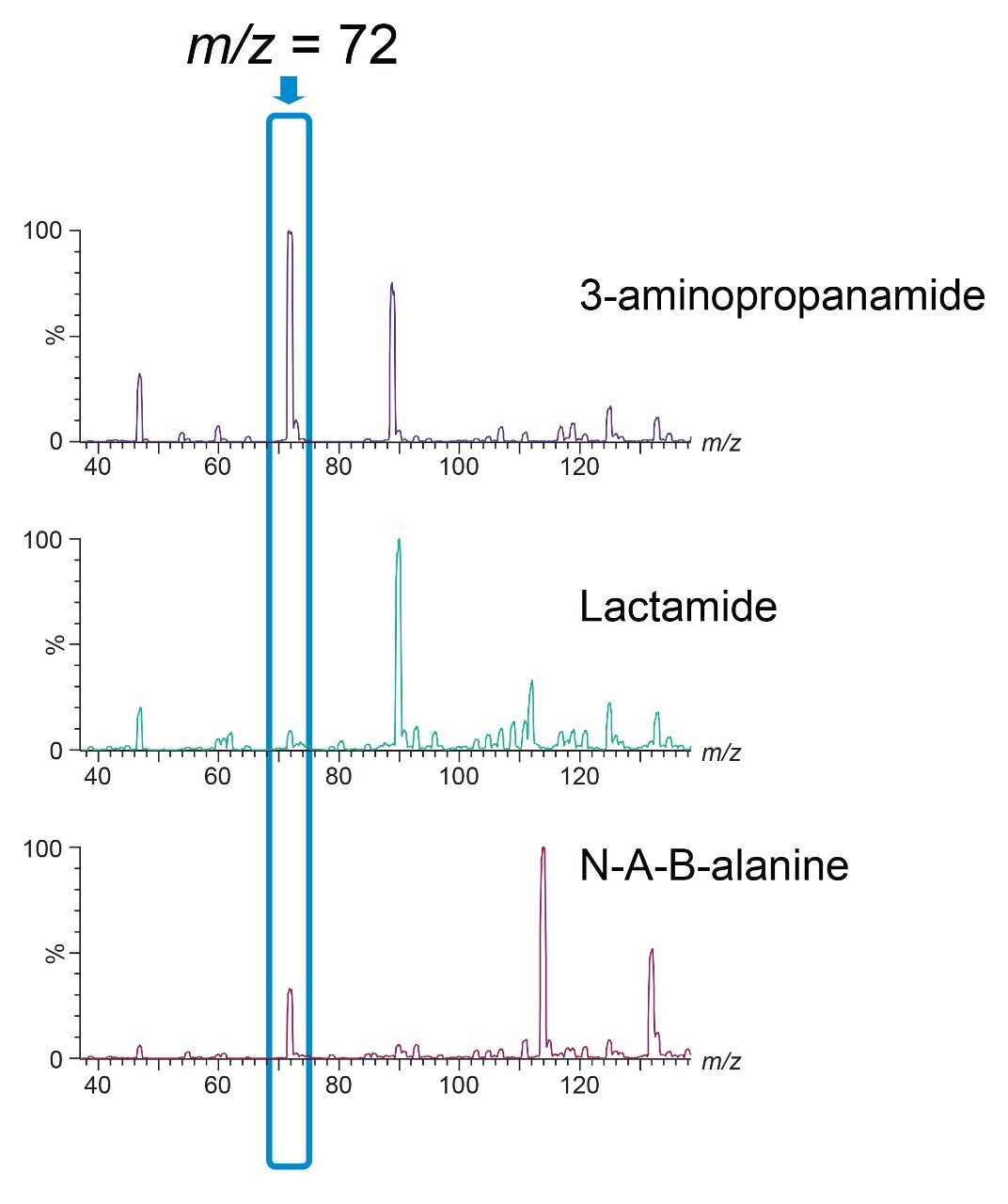
A recent study has identified the presence of other compounds that can interfere with the quantification of acrylamide in various food matrices such as coffee.4 Potential isobaric impurities of acrylamide were studied and an in-source fragment of N-acetyl-β-alanine was identified as the main isobaric ion. 3-aminopropanamide and lactamide were some of the other notable interreferences to be reported. Although our methodology had gone through vigorous testing as part of the validation, in light of these recent findings regarding possible isobaric interference, we re-investigated the selectivity of our method.