Routine Determination of Per- and Polyfluorinated Alkyl Substances (PFAS) in Drinking Water by Direct Injection Using UPLC-MS/MS to Meet the EU Drinking Water Directive 2020/2184 Requirements
Abstract
The purpose of this work is to demonstrate a direct injection UPLC-MS/MS method for the determination of PFAS compounds in drinking water in response to the parametric level requirements in the 2020 recast EU Drinking Water Directive.1 The method performance study was completed on an ACQUITY UPLC I-Class PLUS System with a Xevo TQ-XS and UniSpray ion source combination using an ACQUITY Premier BEH Shield RP18 Column. Samples were prepared by dilution with an acidified organic solution containing internal standard directly into an autosampler vial.
A method validation study was carried out on 3 common drinking water matrices; tap water from known soft and hardwater areas and bottled mineral water. The method performance was assessed using 3 spike levels at 2, 10, and 100 ng/L (1.2, 6, and 60 ng/L in vial concentration) for all analytes, with 7 replicates at each level. Twelve analytes were corrected using internal standards with no suitable internal standard for PFPS, PFHpS, PFNS, PFTrDA, PFDS, PFUnDS, PFDoDS, and PFTrDS. Average method performance for trueness was 89 to 112% across all matrices. RSDs were all at or below 14%.
All calibration graphs had residuals within 20% and R2 values of 0.99 or higher with linear regression fit. Retention time stability across all the method validation study batches for all analytes was below RSD 3.2%.
Benefits
- Sensitive analysis to determine residues at concentrations as low as 1 ng/L in drinking water without the need for lengthy cleanup or concentration steps
- Confidence in results by reducing and separating possible system and solvent contaminants with the utilization of the Waters PFAS kit and isolation column for LC modification
- Provides a total solution using a direct injection, UPLC-MS/MS method suitable for the detection and quantification of all 20 PFAS with an LOQ of 1 ng/L per compound which significantly exceeds requirements in the 2020 EU Drinking Water Directive
Introduction
Due to their unique properties, diverse array of functional groups, and stability, per- and polyfluoroalkyl substances (PFAS) are extremely useful and widely used in industry. As a result, these persistent and bio-accumulative pollutants are commonly found in a large range of consumer goods and by-products of industrial processes and are introduced to the environment through a variety of sources ranging from the manufacturing of non-stick coatings to their use in firefighting foams.
However, the chemical properties that make them a desirable commodity also make them non-biodegradable, easily mobile within different environmental compartments, and extremely persistent in the environment and our bodies. Studies have linked them to adverse effects on human health such as affecting immune and developmental systems as well as being possible carcinogens.
The EU has now regulated a set of 20 PFAS compounds in the updated European Drinking Water Directive, 2020/2184.1 This acknowledges the high contamination potential of PFAS compounds and imposes a limit of 0.1 µg/L for the sum of 20 specified individual PFAS compounds that are considered a concern in water intended for human consumption. This application note demonstrates the performance of the ACQUITY UPLC I-Class PLUS coupled with a Xevo TQ-XS with three drinking water matrices: soft tap water, hard tap water, and mineral water. These samples were diluted with an acidified organic solution (containing internal standard) and directly injected into the LC-MS/MS without additional concentration or clean-up required.
The detection requirement for the 20 individual PFAS compounds are in the ng/L, or parts per trillion (ppt), range which requires a sensitive analysis method. PFAS respond well using negative electrospray ionization (ESI-) and this has become the preferred method of analysis. Although this ionization technique works well, the increasingly stringent requirement for detection can benefit from any increase in response that a technique like UniSpray could provide.
UniSpray is a novel atmospheric ionization technique that allows for multimode ionization of both polar and non-polar analytes in a single injection. The column effluent is nebulized as it exits a grounded, heated probe. The spray is directed onto a stainless-steel pin that is held at high voltage. This creates similar droplets to electrospray, leading to increased desolvation of ions. The nebulized flow bends around the surface of the impactor pin into the sample cone due to the Coanda effect. This mechanism allows for increased ionization and sampling efficiency.2-3
As an added investigation into further improving sensitivity, an ACQUITY Premier Column with equivalent chemistry to a traditional ACQUITY UPLC Column was assessed in a comparison study. ACQUITY Premier Columns, featuring MaxPeak High Performance Surfaces (HPS) Technology, are designed to maximize separation performance, and reproducibility by minimizing analyte/flow path interactions that can lead to analyte losses and poor chromatographic performance.
Experimental
Sample Description
One liter of water was collected from sources of known soft and hard water areas in the UK and stored in 50 mL centrifuge tubes at room temperature until analysis. Mineral water was purchased from a UK retail outlet and stored at room temperature in its original container before use.
Because of the widespread use of PFAS, required detection limits are in the low ng/L range and specific challenges must be addressed for sample collection, preparation, and analysis. In the lab, items to avoid due to contamination potential include sticky notes, certain disposable pipettes, vial caps with Teflon seals, and LDPE containers, to name a few. It is recommended that where practical, screening of consumables and reagents for the presence of PFAS should be made before the analysis of samples.
Contamination is unavoidable from the chromatographic system and solvents. Therefore, steps should be taken to minimize these contributions, and as such, the PFAS kit was installed which is comprised of PFAS-free components that replaces items such as the conventional Teflon coated solvent lines with PEEK tubing. An isolator column was also installed which helps to delay any residual background interferences from co-eluting with the analytical peak. Installation of both the kit and column is straightforward and quick.4
Method Conditions
300 µL water sample was aliquoted directly into an autosampler vial, diluted with 200 µL acidified organic reagent containing 10 ng/L internal standard and sealed for direct injection by UPLC-MS/MS. See Figure 1 for preparation workflow for samples. The ratio of components in the final sample was the equivalent to 60:29.9:10:0.1 v/v/v/v water sample:acetonitrile:methanol:formic acid.
 Figure 1. Preparation workflow for drinking water samples.
Figure 1. Preparation workflow for drinking water samples.
Recovery spikes were carried out on all 3 matrices with 7 replicates at each level at 2 ng/L, 10 ng/L, and 100 ng/L for all 20 PFAS written into the 2020 EU Drinking Water Directive. Matrix matched standards were prepared in the respective blank extracts. The calibration range was 1 to 200 ng/L for all analytes. Quantification of spiked samples was by matrix matched bracketed calibration using internal standards where available, see Appendix A for details as suitable internal standards were not available for all PFAS analytes. The MRMs listed in Appendix A were used in this application for quantification and confirmation of residues.
LC Conditions
|
LC system: |
ACQUITY UPLC I-Class PLUS with fixed-loop Sample Manager fitted with Waters PFAS kit (p/n: 205000588) and isolator column (p/n: 186004476) |
|
Detection: |
Xevo TQ-XS |
|
Vials: |
Polypropylene 12 x 32 mm Screw Neck Vial, with polyethylene septumless cap, 700 µL (p/n 186005230) |
|
Column: |
ACQUITY Premier BEH Shield RP18, 1.7 µm, 2.1 mm x 100 mm (p/n: 186009498) |
|
Column temp.: |
45 °C |
|
Sample temp.: |
15 °C |
|
Injection volume: |
50 µL |
|
Flow rate: |
0.350 mL/min |
|
Mobile phase A: |
2 mM ammonium acetate in water: methanol 95:5 (v/v) |
|
Mobile phase B: |
2 mM ammonium acetate in methanol |
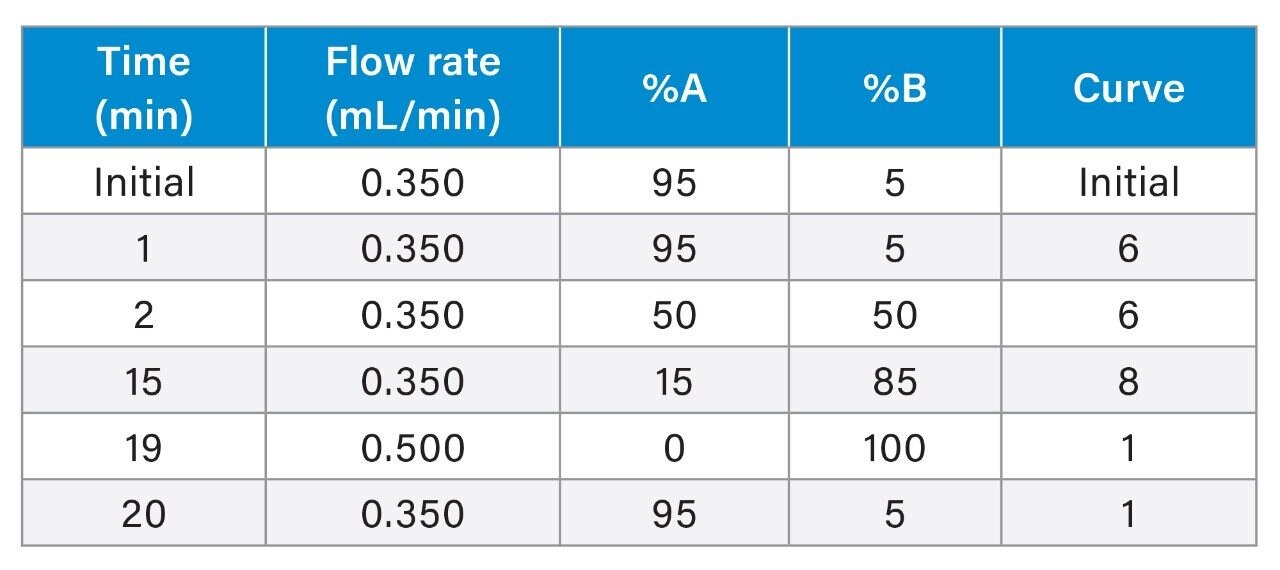
Gradient Table
MS Conditions
|
MS system: |
Xevo TQ-XS |
|
Ionization mode: |
UniSpray negative |
|
Acquisition range: |
MRM |
|
Impactor voltage: |
0.9 kV |
|
Desolvation temperature: |
400 °C |
|
Desolvation gas flow: |
1000 L/hr |
|
Cone gas flow: |
150 L/hr |
|
Source temperature: |
110 °C |
MRM Transitions
MRM parameters for each compound are listed in Appendix A.
Data Management
|
Informatics: |
MassLynx v4.2 |
Results and Discussion
Effect of sample composition on peak shape
When analyzing short and long chain PFAS compounds simultaneously, a careful balance in final sample composition is required to maximum performance of all analytes. Subsequently, a reduction in sensitivity was observed for the less water-soluble longer chain PFAS compounds when diluting with an increasingly aqueous composition. After reducing aqueous content to address solubility of long chain PFAS, the final sample composition was stronger in organic content than the initial LC starting conditions, causing peak broadening of the early eluting short chain compounds. Final sample composition in relation to the LC starting conditions was carefully considered, particularly in the case when higher injection volumes were used.
A range of compositions varying in the composition of aqueous sample to organic solvent was studied. Methanol and acetonitrile were both independently investigated, as well as a mixture of the two as the organic component of the dilution. A mixture of acetonitrile and methanol was found to be the best diluent for the aqueous sample to optimize compounds over the whole spectrum of PFAS chain lengths included in the analysis. Examples of this are given in Figure 2. Acetic acid was initially used to acidify the samples to increase gains in sensitivity, but the reagent contained trace levels of PFAS and was replaced with formic acid. Diluting the sample directly in vial helped to mitigate any compound loss.
To further improve the peak shape of early eluting compounds, an extension loop assembly (50 µL, p/n: 430002012) and ACQUITY Column In-Line Filter Kit (p/n: 205000343) was installed between the injector valve and the analytical column. This provides extra void volume prior to sample loading onto the column to allow additional sample/mobile phase mixing, providing analyte focusing on the LC column.
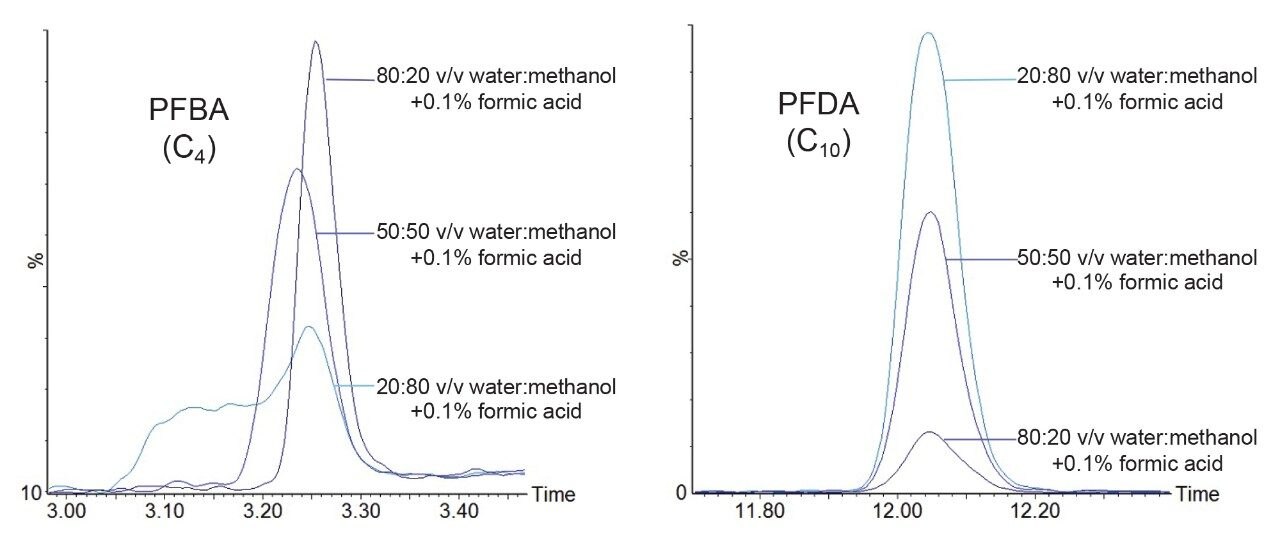 Figure 2. Effect of sample composition on sensitivity and peak shape on short chain and longer chain PFAS.
Figure 2. Effect of sample composition on sensitivity and peak shape on short chain and longer chain PFAS.
Advantages of Using ACQUITY Premier Columns for Longer Chain PFAS
Initial method development applied the use of an ACQUITY UPLC BEH Shield RP18 Column which achieved suitable peak shape and separation. An additional investigation into the ACQUITY Premier BEH Shield RP18 Column was conducted to explore any potential enhancements this could give for the analysis. For the short-to-mid chain compounds there was no significant change in sensitivity using the ACQUITY Premier Column compared to the ACQUITY UPLC Column. However, the longer chain compounds (≥C10) displayed a significant gain in sensitivity; up to 20 times the area and signal to noise at the longest chain length PFAS tested, PFTrDS. As the longer chain PFAS were substantially challenging in terms of sensitivity given the required final sample composition, the enhancements identified with the ACQUITY Premier BEH Shield RP18 Column demonstrated why this column was selected for this application. Example chromatograms comparing short, mid, and long length chain PFAS using ACQUITY UPLC and ACQUITY Premier Columns are displayed in Figure 3.
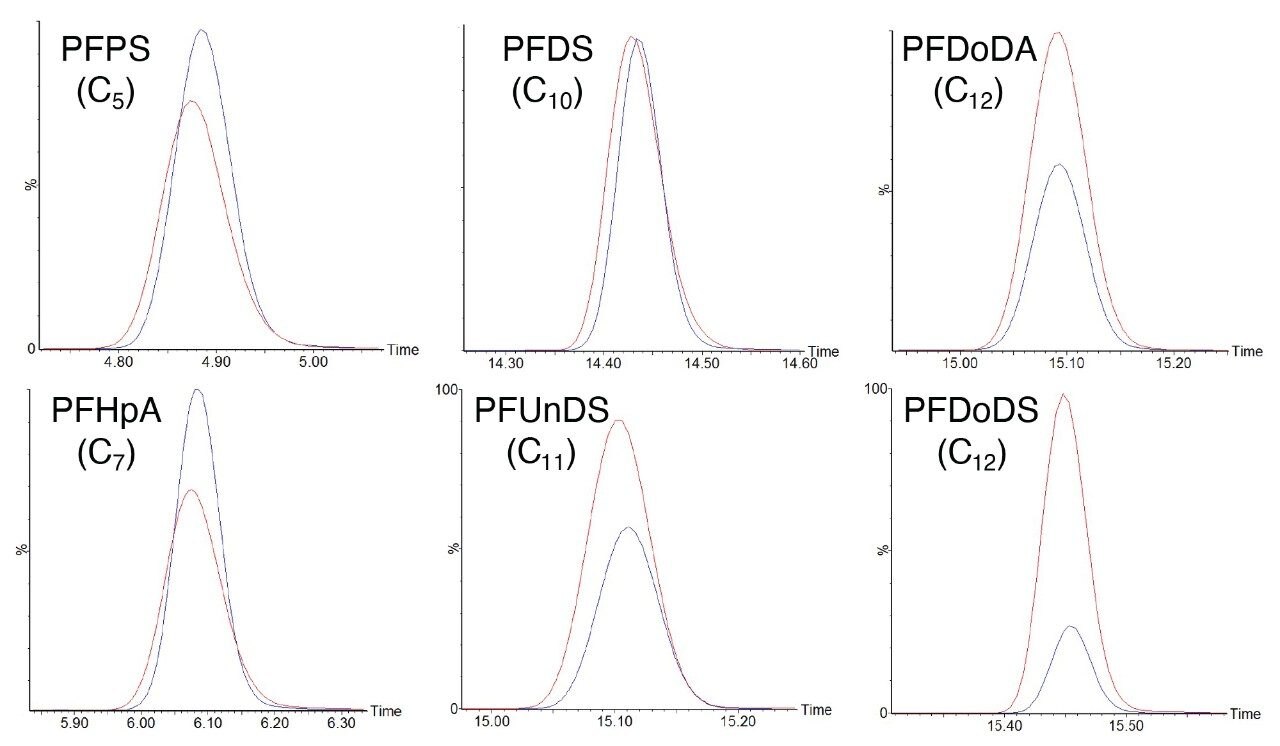 Figure 3. Peak response of PFAS compounds comparing ACQUITY Premier Column (red) and ACQUITY UPLC Column (blue) performance over a range of chain lengths. Demonstrated at 10 ng/L in LCMS grade water.
Figure 3. Peak response of PFAS compounds comparing ACQUITY Premier Column (red) and ACQUITY UPLC Column (blue) performance over a range of chain lengths. Demonstrated at 10 ng/L in LCMS grade water.
Improving Detection and Sensitivity Through Use of the UniSpray Ion Source
Direct comparison by electrospray and UniSpray ionization was made by repeating the validation batch containing hard water and LCMS grade water samples on each source type. The UniSpray ion source displayed consistent gains in sensitivity for all compounds, measured by comparing peak area, peak height, and signal to noise. Comparison chromatograms are displayed in Figure 4. An average increase of 18 times in peak area and 5 times in signal to noise ratio was observed. By improving the detection sensitivity for the long chain PFAS the injection volume could be adjusted to a suitable volume that would improve chromatography in the early eluting short chain PFAS. A final injection volume of 50 µL was determined to provide adequate sensitivity with suitable peak shape.
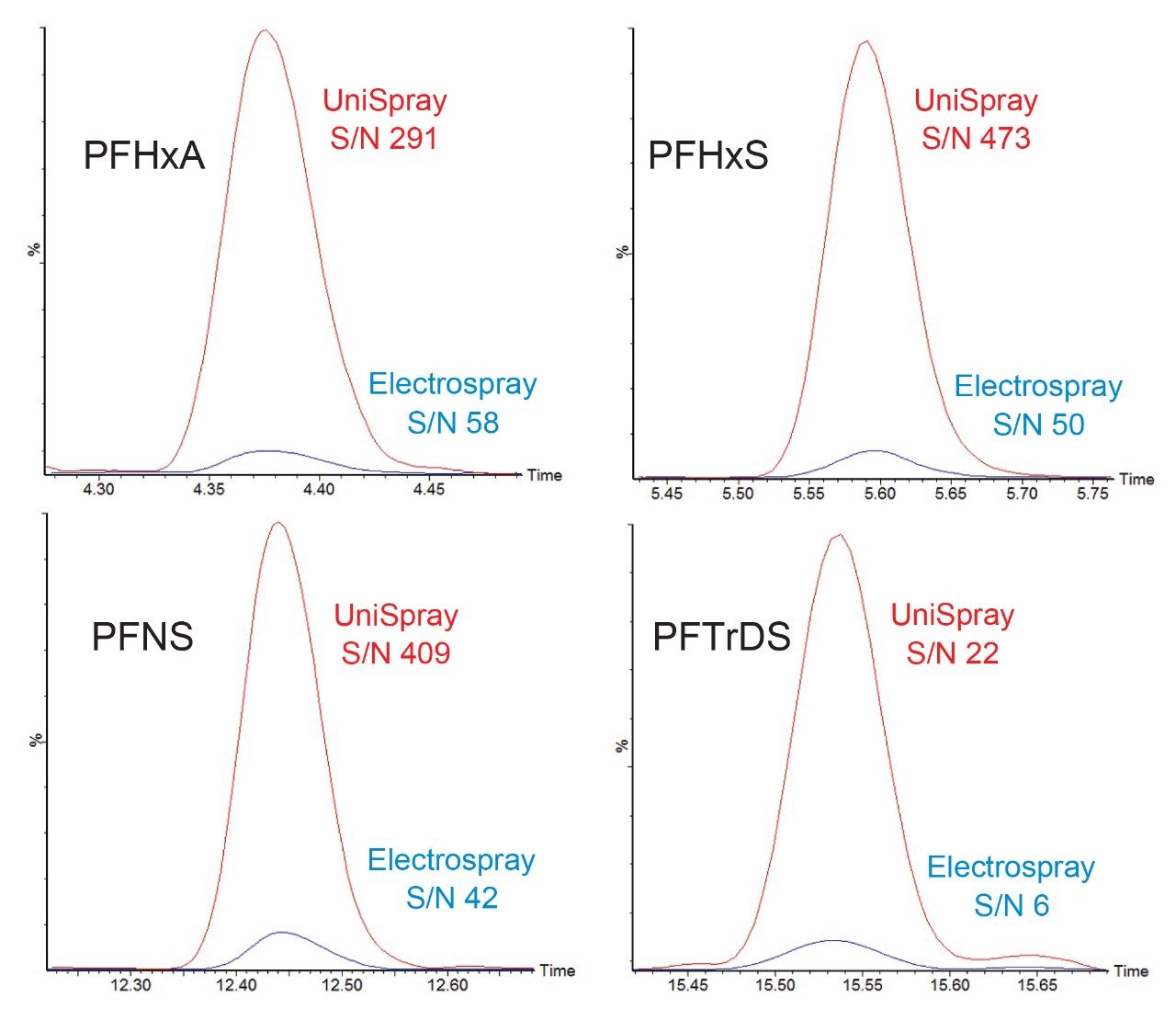 Figure 4. Signal to noise and peak response comparison between UniSpray (red) and Electrospray (blue) ionization techniques demonstrated at 2 ng/L in hard water (average S/N).
Figure 4. Signal to noise and peak response comparison between UniSpray (red) and Electrospray (blue) ionization techniques demonstrated at 2 ng/L in hard water (average S/N).
Method Validation Study Results
Matrix matched calibration was used throughout the method validation study and example calibration curves are demonstrated in Figure 5. For all analytes, each calibration graph in the validation study displayed coefficients of determination of 0.990 or higher using a 1/X linear regression fit, with residuals all <20%. Figure 5 also shows chromatograms for compounds injected at 1 ng/L concentration in water.
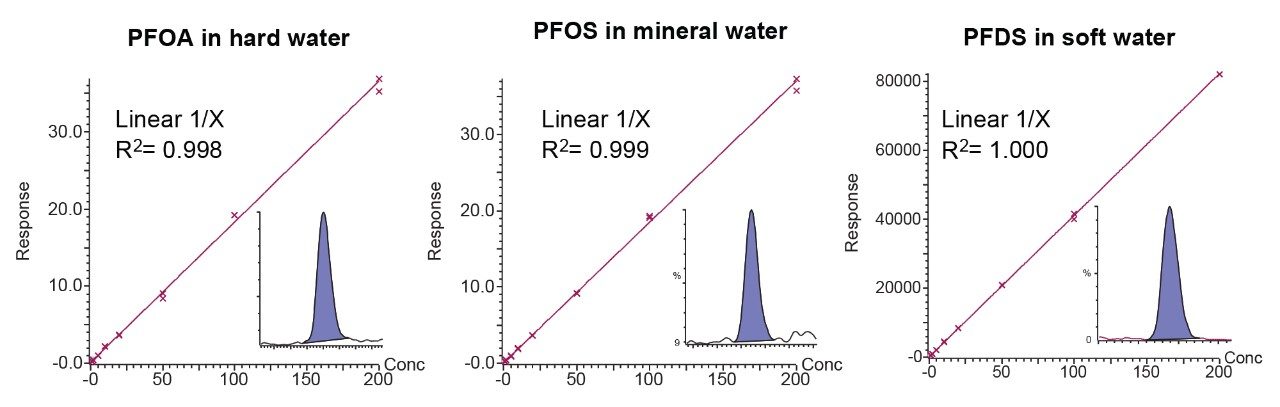 Figure 5. Bracketed matrix-matched calibration curves for a selection of PFAS at 1 ng/L to 200 ng/L (0.6 ng/L to 120 ng/L in vial concentration), including chromatograms for the quantitative transitions at 1 ng/L. All residuals are within 20% of nominal values.
Figure 5. Bracketed matrix-matched calibration curves for a selection of PFAS at 1 ng/L to 200 ng/L (0.6 ng/L to 120 ng/L in vial concentration), including chromatograms for the quantitative transitions at 1 ng/L. All residuals are within 20% of nominal values.
Overall, method performance can be summarized in the recovery values highlighted in Figure 6 which was assessed over three validation batches and covered the following typical drinking water types with a range of properties: soft, hard, and mineral water. Each of these batches contained 7 spiked recoveries at three levels: 2, 10, and 100 ng/L. All independent PFAS compounds, levels and matrices gave an average recovery by compound between 80 and 120% and is displayed in Figure 6. Repeatability of the method was assessed from the recovery samples and all PFAS had an RSD below 13% with the exception of the longest chain sulfonate, PFTrDS, in soft water at 23.5%. The percentage RSD values are summarized by error bars in Figure 6.
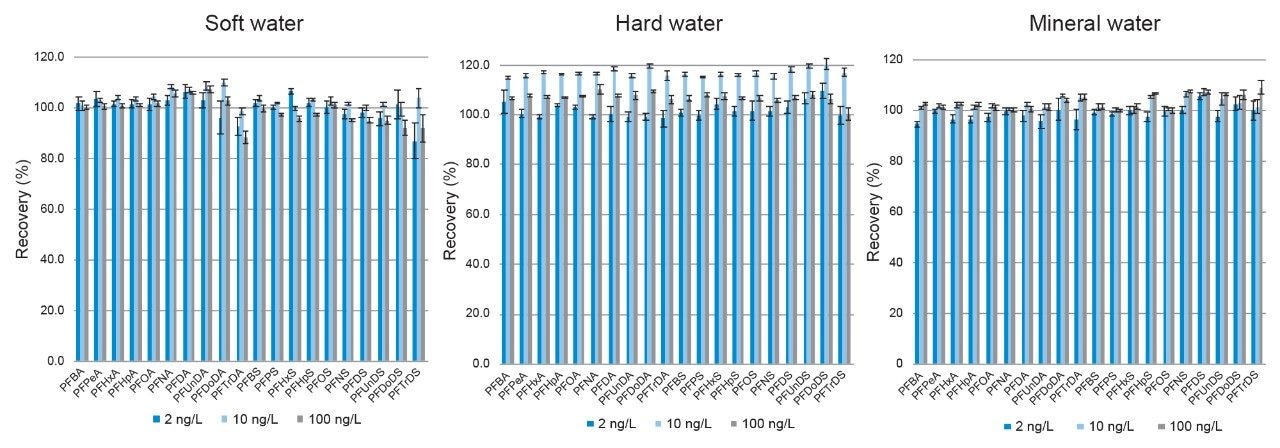 Figure 6. Recovery of 20 PFAS included in method for soft, hard, and mineral water fortified at 2, 10, and 100 ng/L. Error bars represent the percentage RSD for each compound across the 7 data points.
Figure 6. Recovery of 20 PFAS included in method for soft, hard, and mineral water fortified at 2, 10, and 100 ng/L. Error bars represent the percentage RSD for each compound across the 7 data points.
Overall retention time for all analytes across the 3 method validation batches was within 3.2% RSD and demonstrated retention time stability over the study regardless of water type. Additional retention time stability tests were conducted on a mineral water sample where 200 injections of a 10 ng/L matrix calibration standard were run without operator intervention. The retention times were stable and retention time RSDs for all compounds across the whole run were within 0.6% RSD with no significant change to the observed peak shape. See Figure 7 for details.
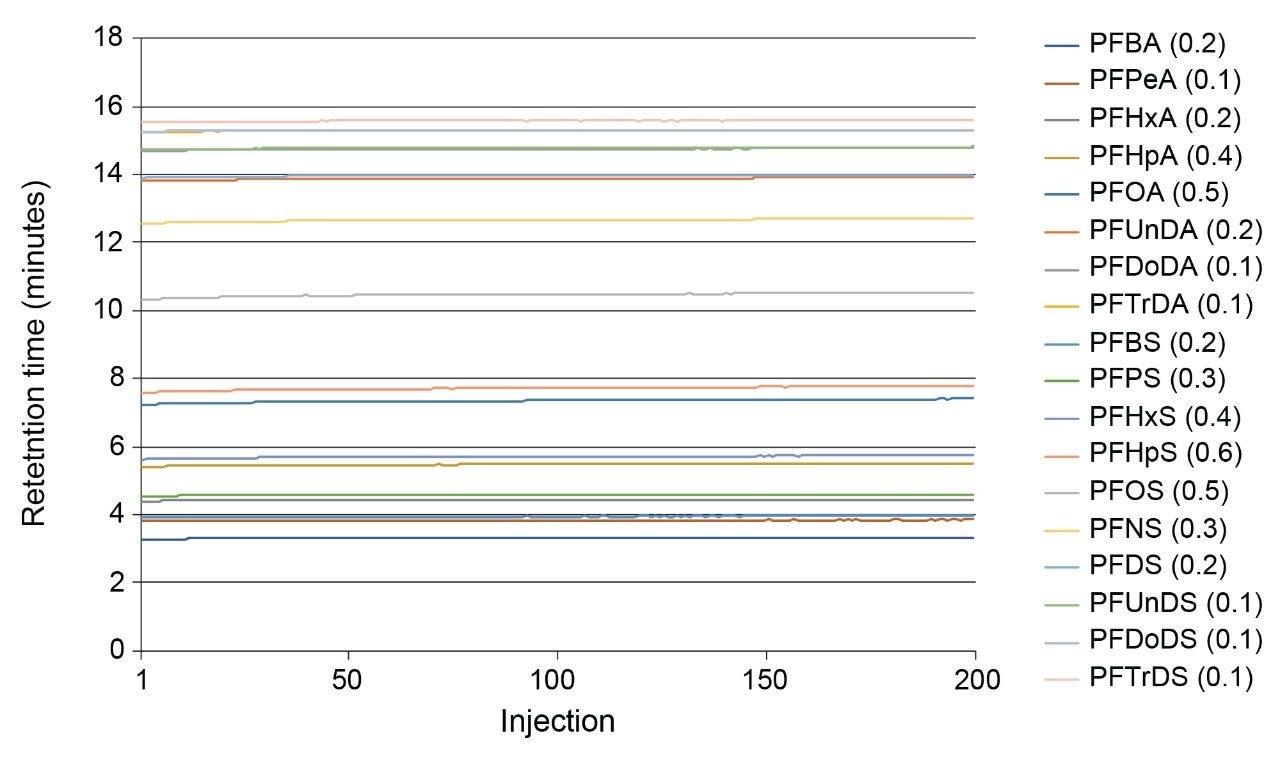 Figure 7. Retention time stability of all included PFAS across 200 injections of mineral water matrix at 10 ng/L. (Percentage RSD values across whole set are given in brackets).
Figure 7. Retention time stability of all included PFAS across 200 injections of mineral water matrix at 10 ng/L. (Percentage RSD values across whole set are given in brackets).
Conclusion
The method validation study results demonstrate a robust analytical method for the determination of Per- and Polyfluoronated Alkyl Substances. The method was optimized to include all 20 PFAS compounds included in the 2020 EU Drinking Water Directive. A low level for each individual PFAS at 1 ng/L was achieved to compliment the 0.1 µg/L sum of PFAS limit required in the monitoring of drinking water. Analysis was performed using a Xevo TQ-XS coupled to an ACQUITY UPLC I-Class PLUS modified with an isolator column and PFAS kit components and separated using an ACQUITY Premier BEH Shield RP18 Column for accurate and reliable results. The use of ACQUITY Premier Column technology and the unique UniSpray ion source enabled achievement of these low concentration levels with a direct injection method without the need for additional clean up or concentration steps.
The trueness and precision of this LC-MS/MS method determined at three matrix QC levels with seven replicate injections was found to be acceptable for all compounds regardless of chain length. Retention time stability and robustness was proven over the course of the study with RSDs for all compounds under 3.2% across all method performance batches. With an LOQ of 1 ng/mL, this method significantly exceeds requirements in the EU Drinking Water Directive 2020/2184.
Scientists must validate the method in their own laboratories and demonstrate that the performance is fit for purpose and meets the needs of the relevant analytical control assurance system.
References
- Directive (EU) 2020/2184 of the European Parliament and of the Council of 16 December 2020 on the Quality of Water Intended for Human Consumption (Recast) [Online] https://eur-lex.europa.eu/eli/dir/2020/2184/oj.
- Lubin A; Bajic S; Cabooter D; Augustijns P; Cuyckens F. Atmospheric Pressure Ionization Using a High Voltage Target Compared to Electrospray Ionization, J. Am. Soc. Mass Spectrom. 2016, 28.
- Lubin A, De Vries R, Cabooter D, Augustijns P, Cuyckens F. An Atmospheric Pressure Ionization Source Using a High Voltage Target Compared to Electrospray Ionization for the LC-MS Analysis of Pharmaceutical Compounds. J. Pharm. Biomed. Anal. 2017, 142.
- Organtini K, Cleland G, Rosnack K. Large Volume Direct Injection Method for the Analysis of Perfluorinated Alkyl Substances (PFAS) in Environmental Water Samples in Accordance with ASTM 7979–17. Waters Application Note, 720006329EN, 2018.
- PFAS Analysis Kit for ACQUITY UPLC Systems. Waters User Guide 715006386, Version 2, 2021.
- Startup Guide for the Analysis of Perfluorinated Alkyl Substances (PFAS) in Environmental Samples. 720006787EN, 2020.
Appendix
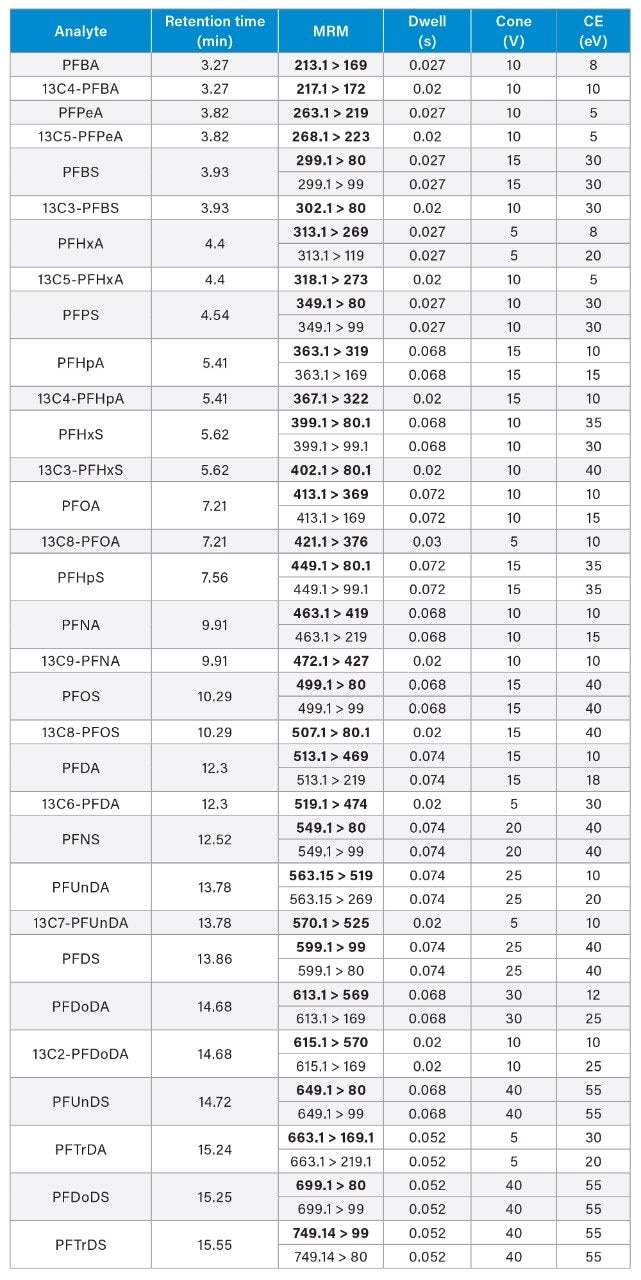 Appendix A. MRM transitions of the analytes and respective isotopically labelled internal standards. Optimum dwell time for target compounds was set automatically using the auto-dwell function (quantitative transitions in bold), so values may vary depending on acquisition windows.
Appendix A. MRM transitions of the analytes and respective isotopically labelled internal standards. Optimum dwell time for target compounds was set automatically using the auto-dwell function (quantitative transitions in bold), so values may vary depending on acquisition windows.
720007413, Revised December 2022