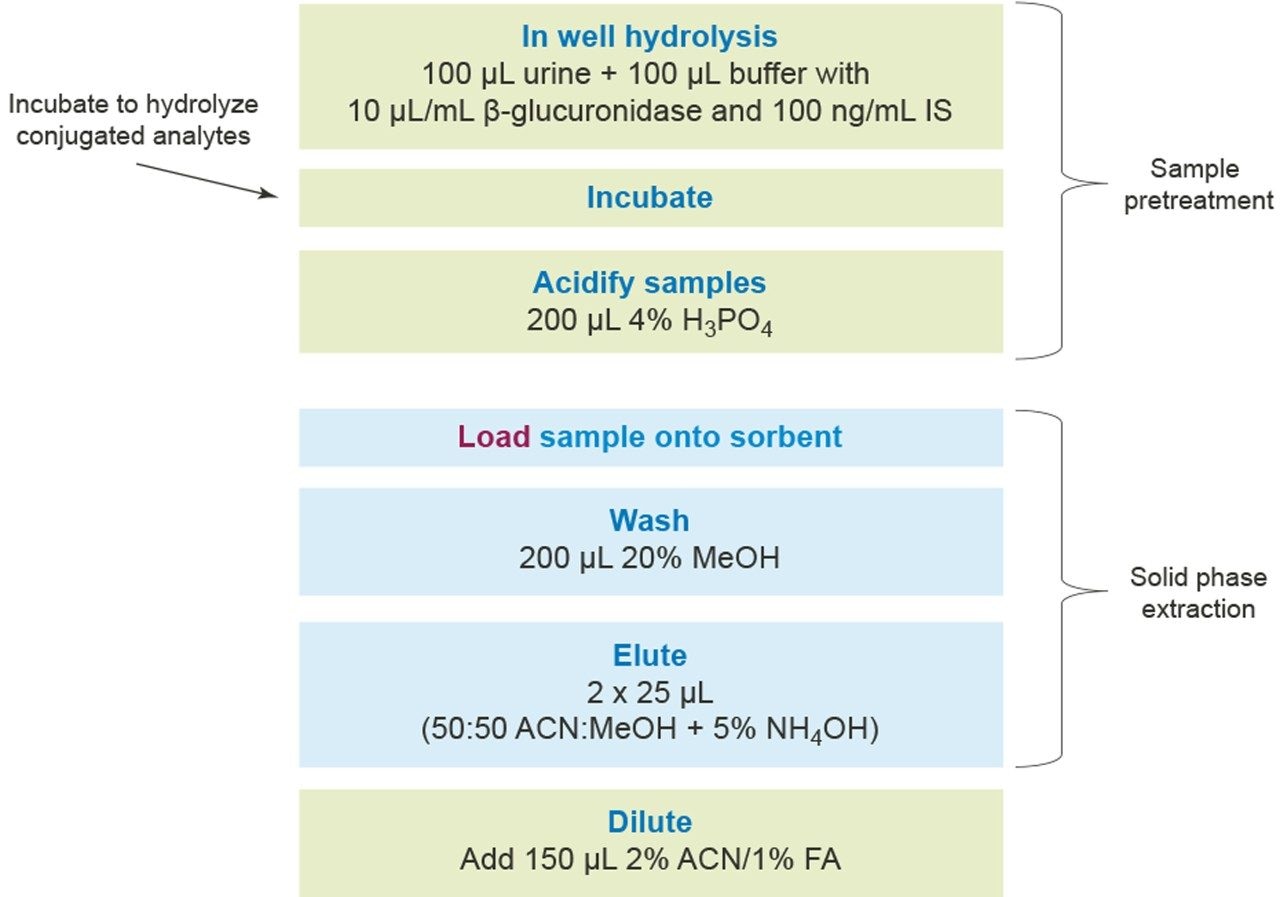
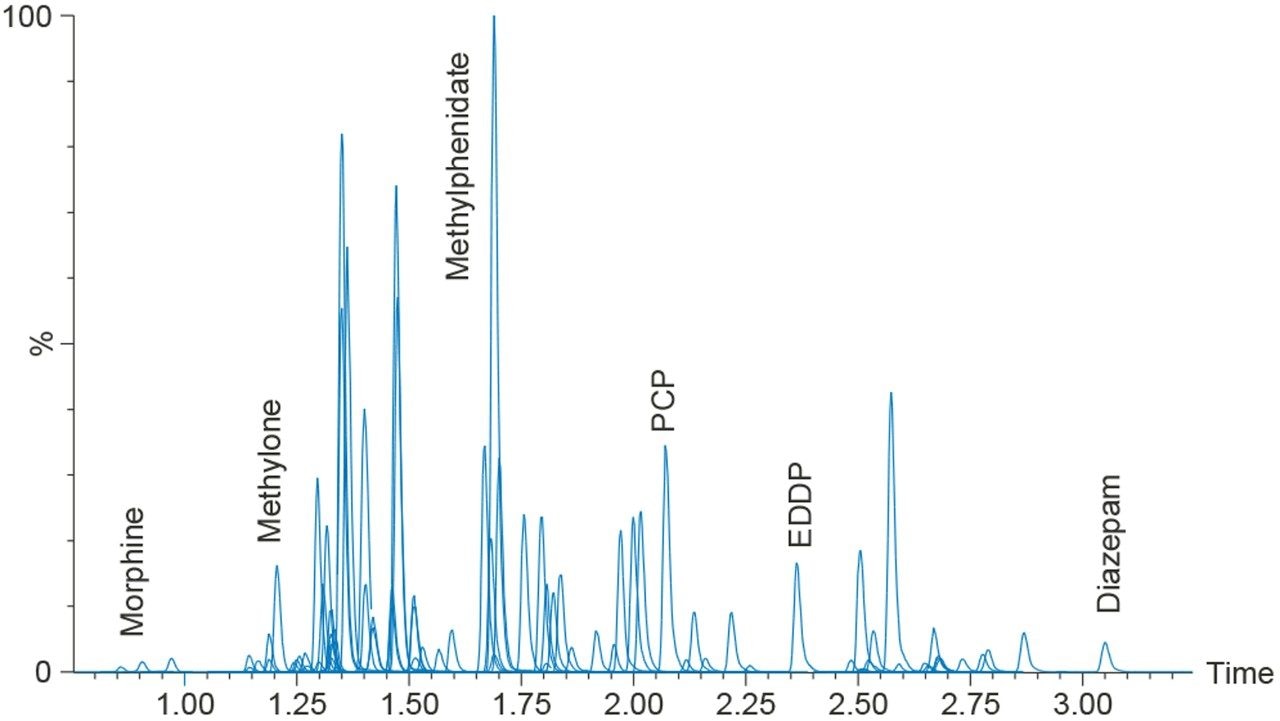
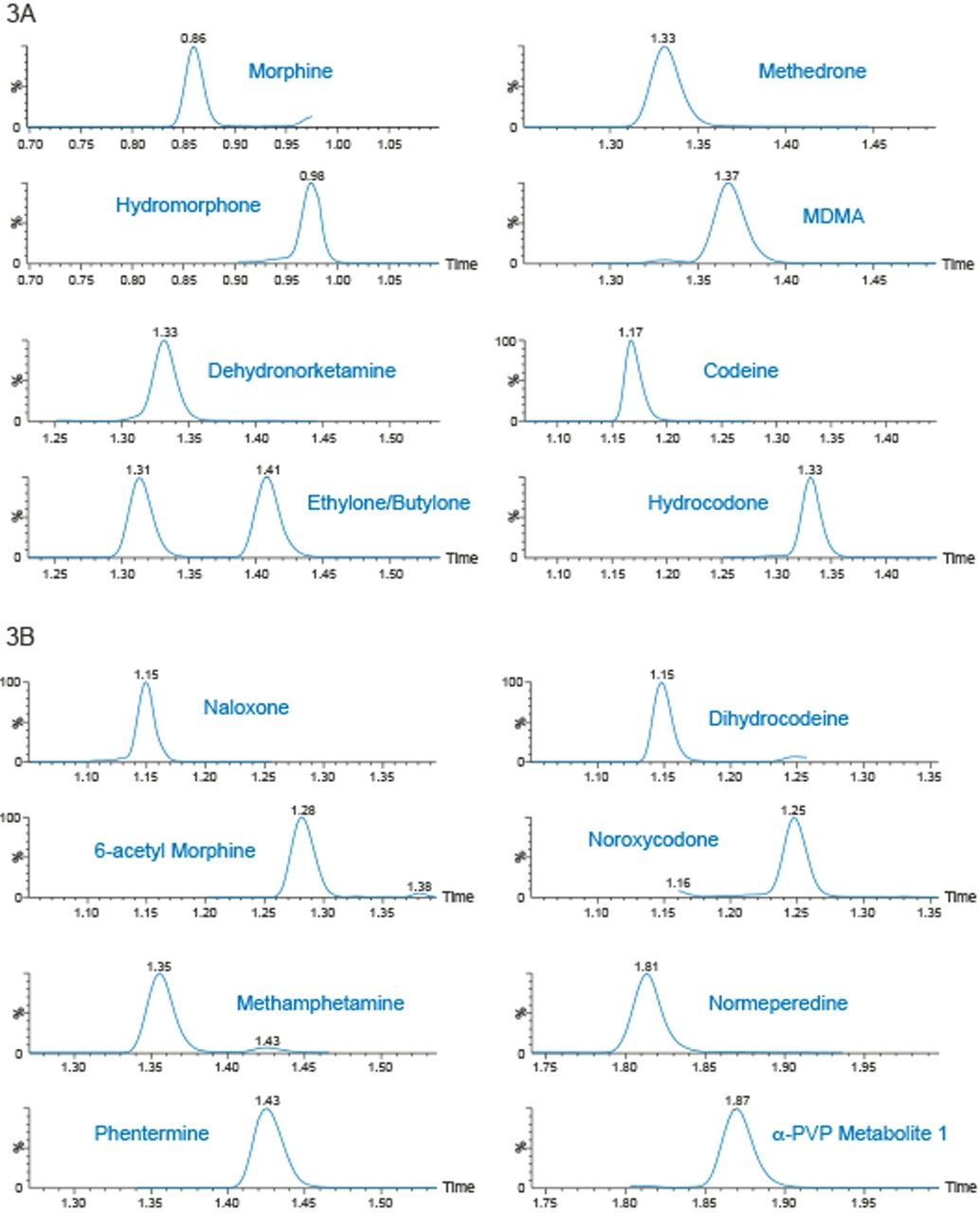
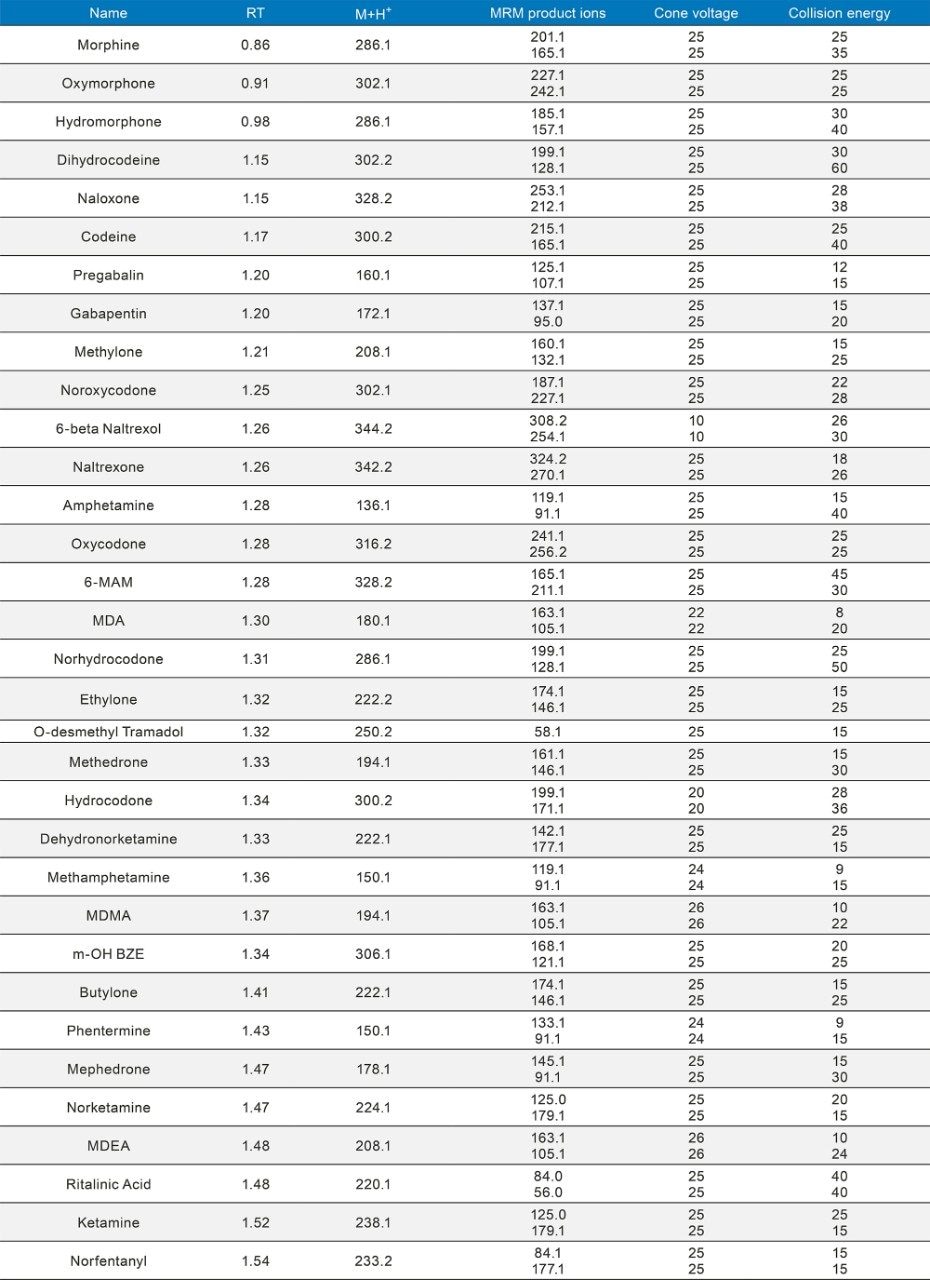
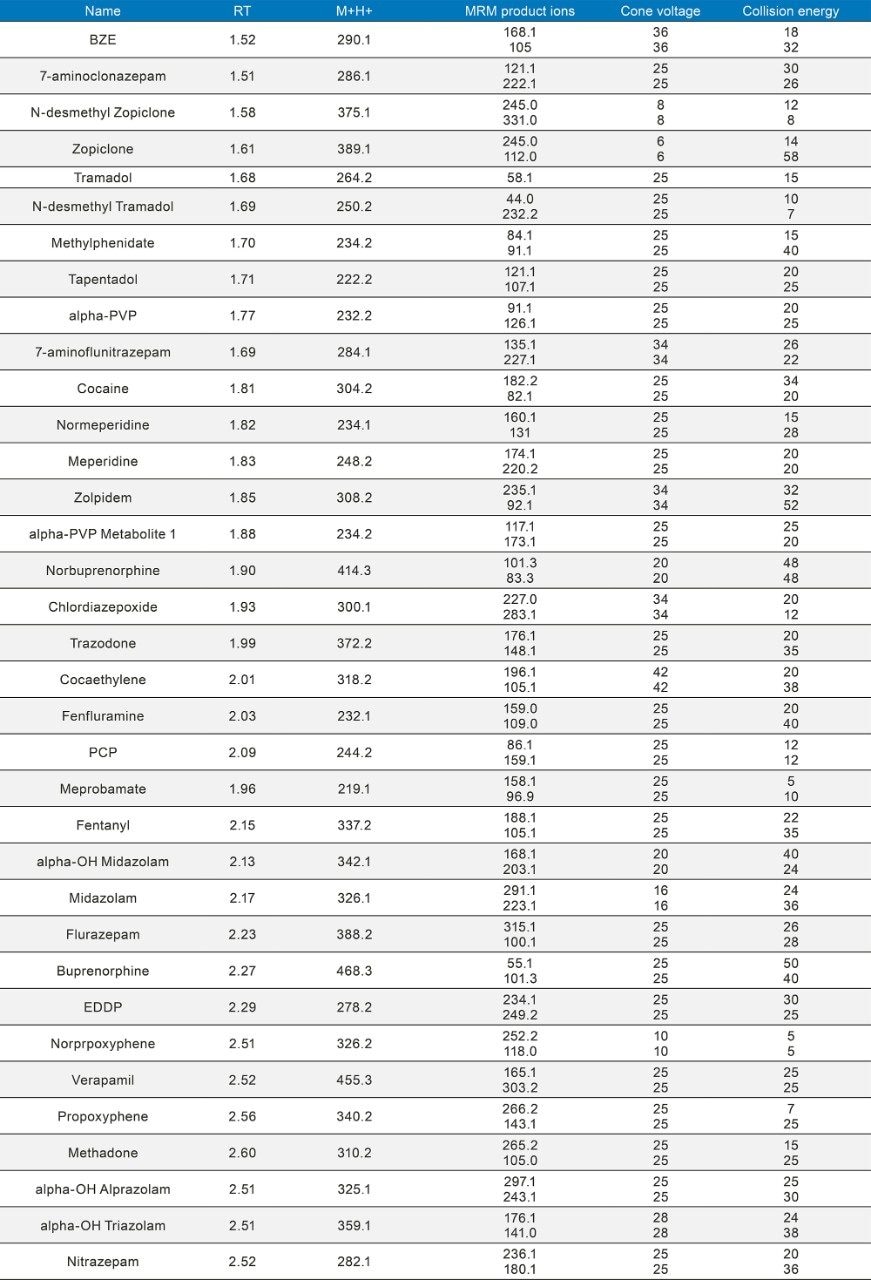
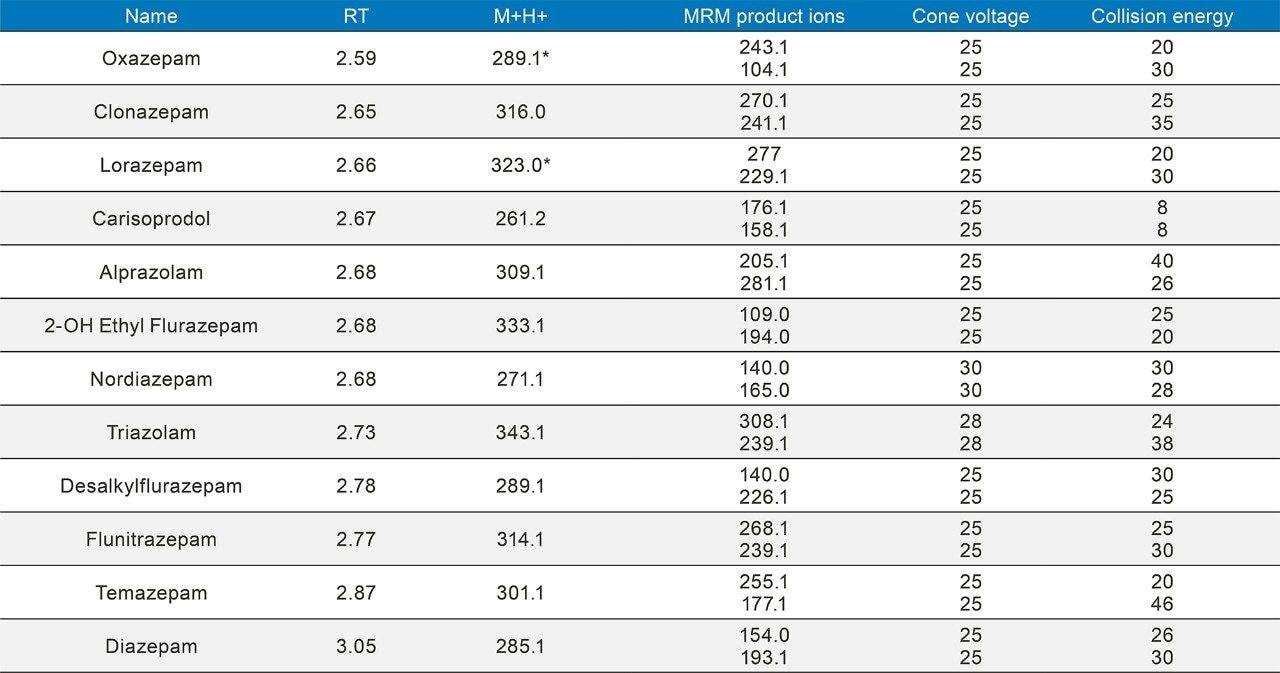
Quantitative Analysis
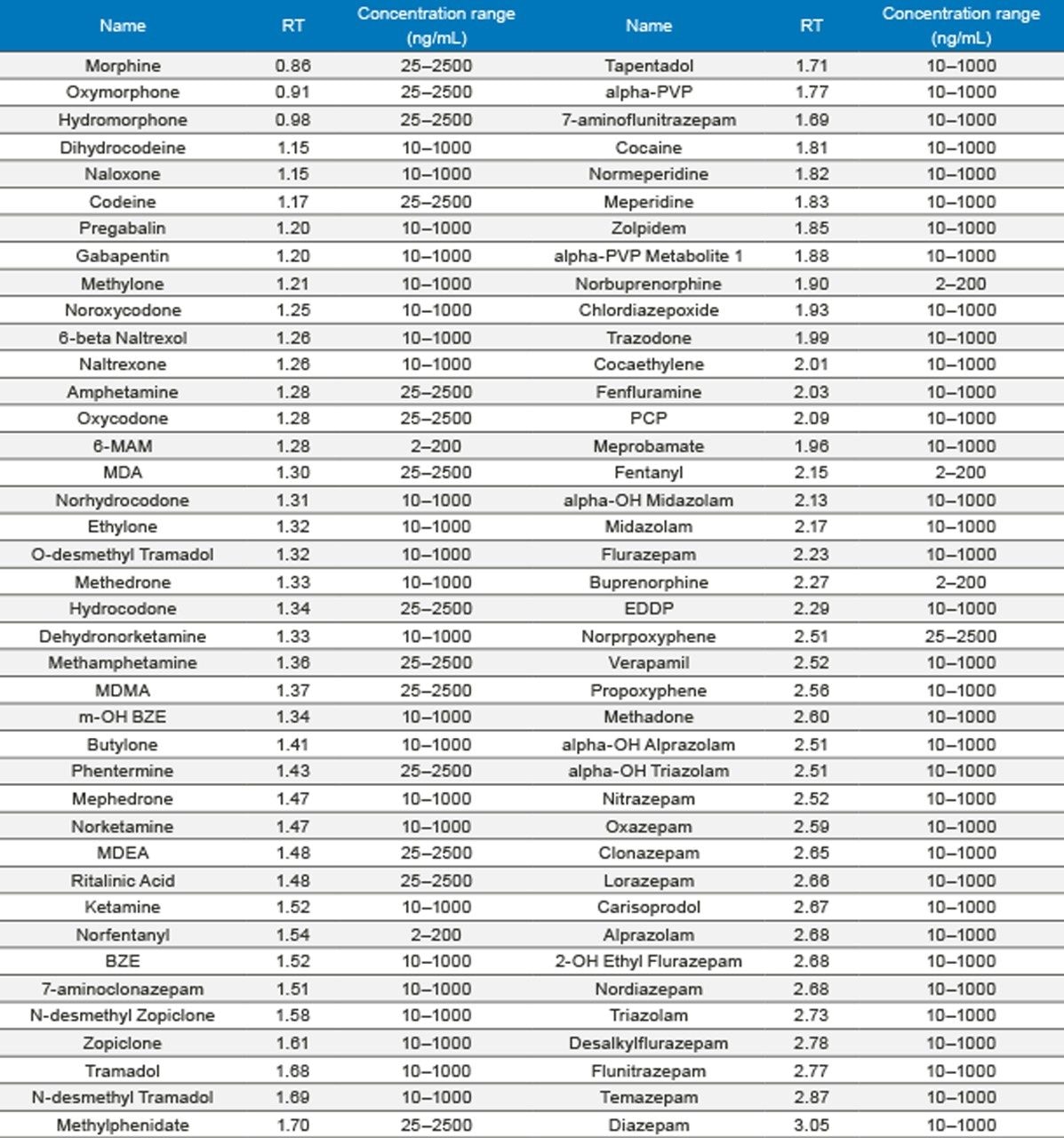
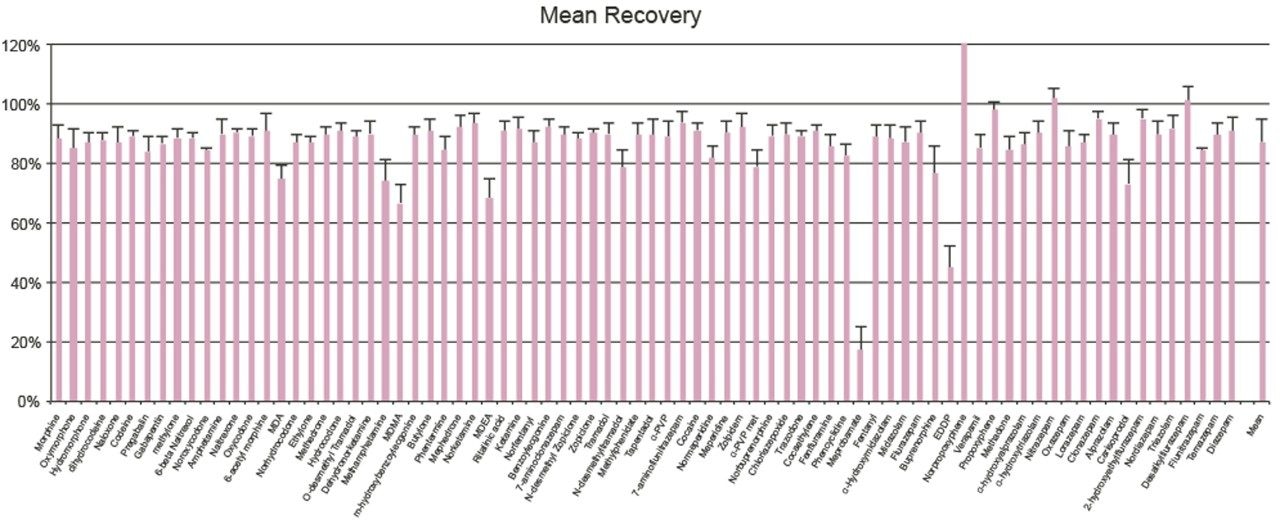
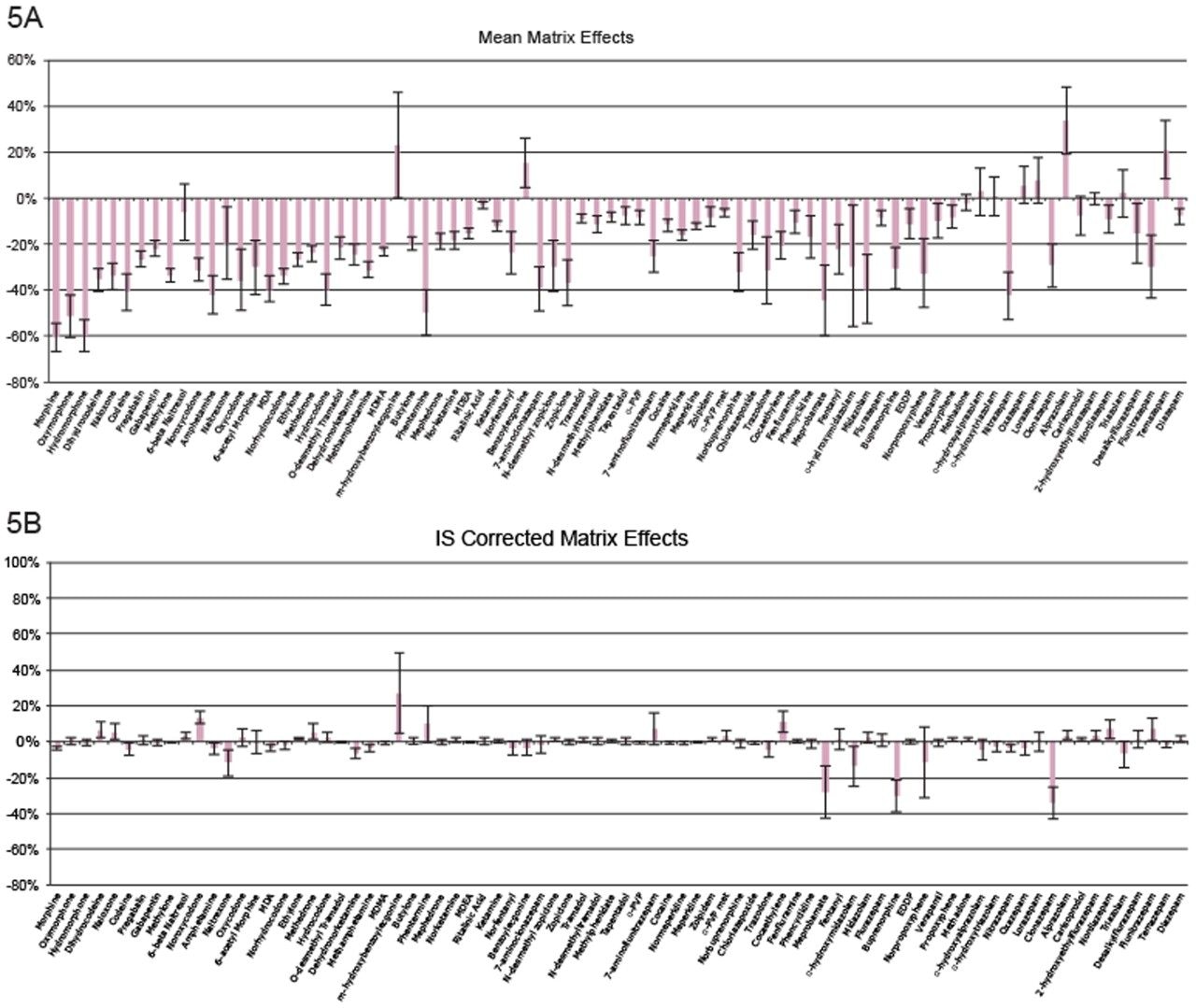
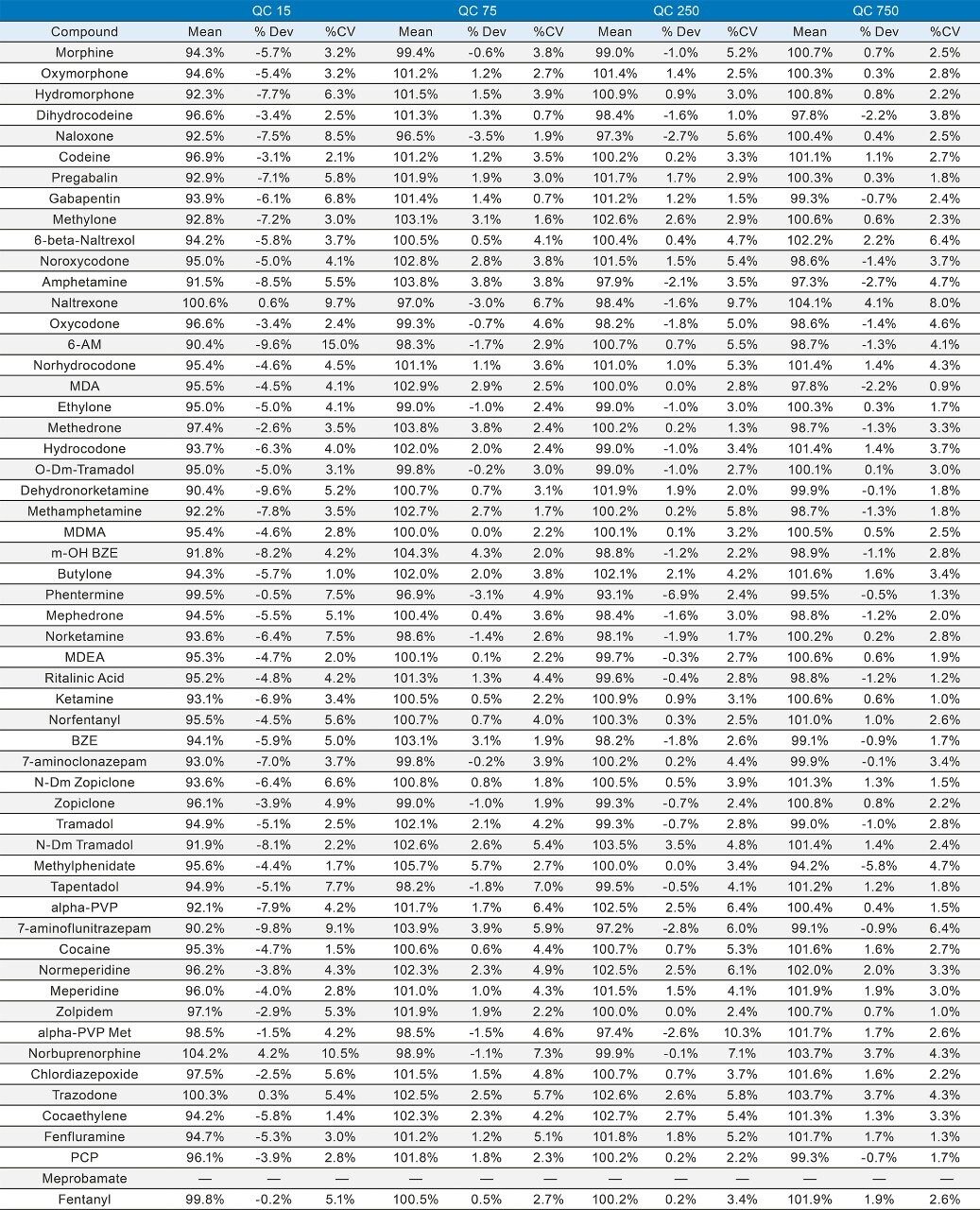
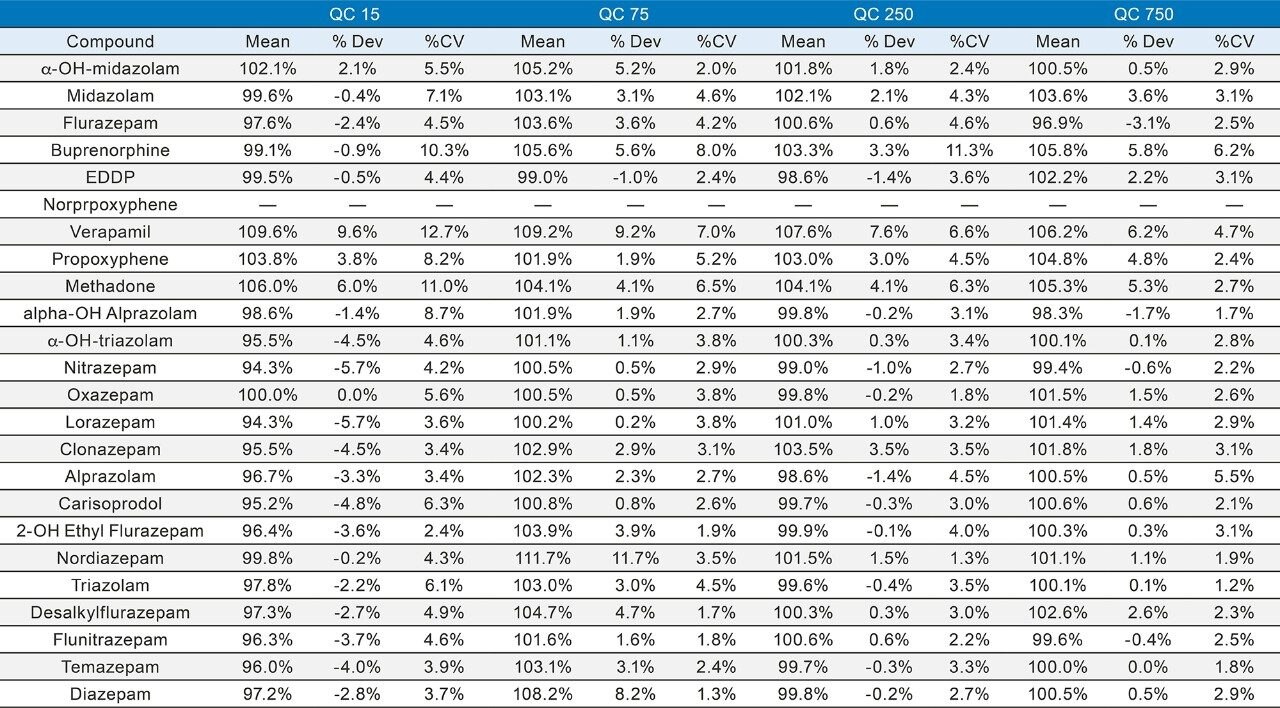
Seven point calibration curves were extracted across the concentration ranges shown in Table 1. Calibration ranges were tailored to reflect the expected concentrations of various compounds. Quality control samples were prepared at 4 concentrations spanning the range of the calibrators, with the lowest at 1.5x the lowest calibrator and the highest at 75% of the highest calibrator. For most compounds, these QC levels were 15, 75, 250, and 750 ng/mL. The compounds at the lower concentrations had QC levels at 3, 15, 50, and 150 ng/mL and the analytes at the higher concentration range had QC levels at 37.5, 187.5, 625, and 1875 ng/mL. Quantitative method validation involved extracting full curves and QC samples over five different days. Calibration curves were extracted in duplicate and six replicates of QC samples were prepared each day. Control limits for individual calibrators and QC samples were ±15% of target values, with the exception of the lowest points, which were required to be within 20%. Precision limits for QC samples were 20% for the lowest QC point and 15% for the other points. Meprobamate and norpropoxyphene were assessed qualitatively only and were not subject to these controls. A summary of the five independent extractions and analyses met all of these criteria and can be seen in Appendix 2. The majority of compounds were within 10% of their target values with %CVs under 10%. For within batch results, all compounds met the accuracy criteria, and the only compound that had precision results greater than 15% was the high amphetamine QC at 18%.
All calibration curves conformed to FDA bioanalytical method validation requirements,3 which dictate that all calibrators be within 15% of target values except the lowest point, which must be within 20% of its target value and that 75% of calibrators meet this criteria. All compounds met these criteria and all curves had R2 values of 0.99 or greater.
Limits of quantification were defined as those points in which the signal was 5X greater than that of an extracted matrix blank, signal to noise ratios were >10, and both bias and %CV were both less than 20%. To evaluate this, six replicates of the lowest calibrator were extracted in one of the validation batches. All compounds met these criteria.
On instrument stability was also assessed. A single batch was extracted and analyzed five times over an eight day period. Through four days, all compounds met the quantitative validation criteria described above.
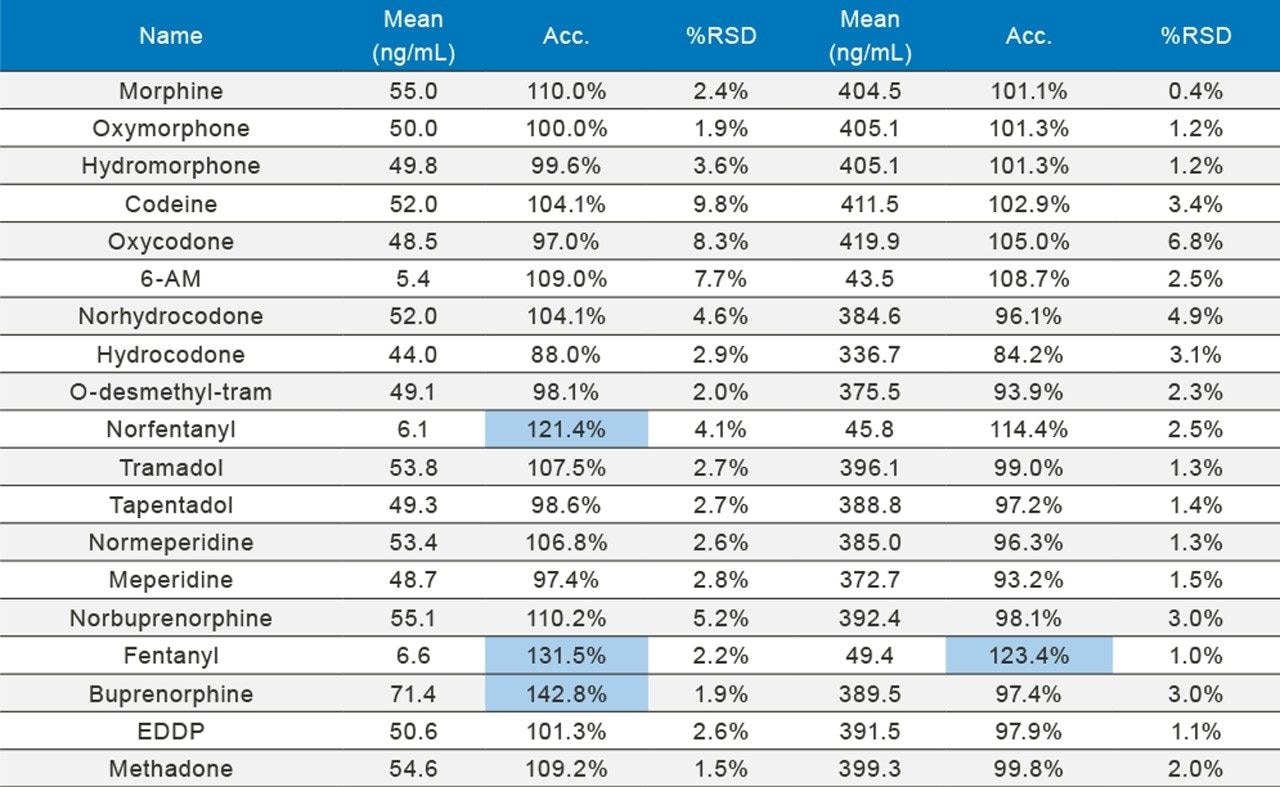
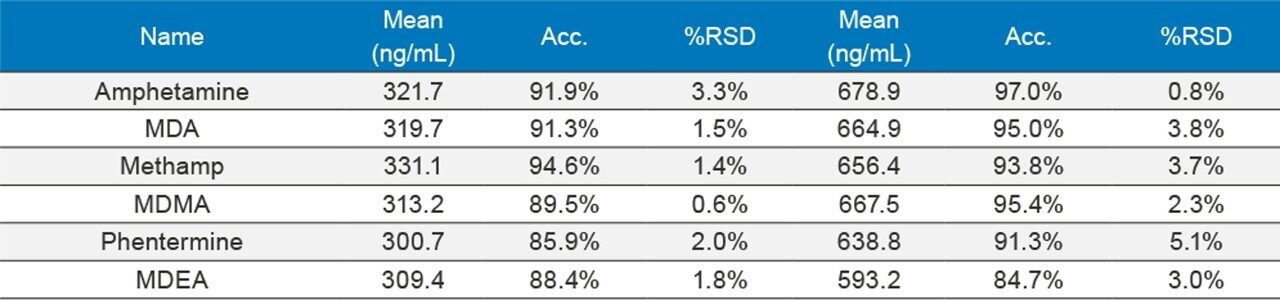
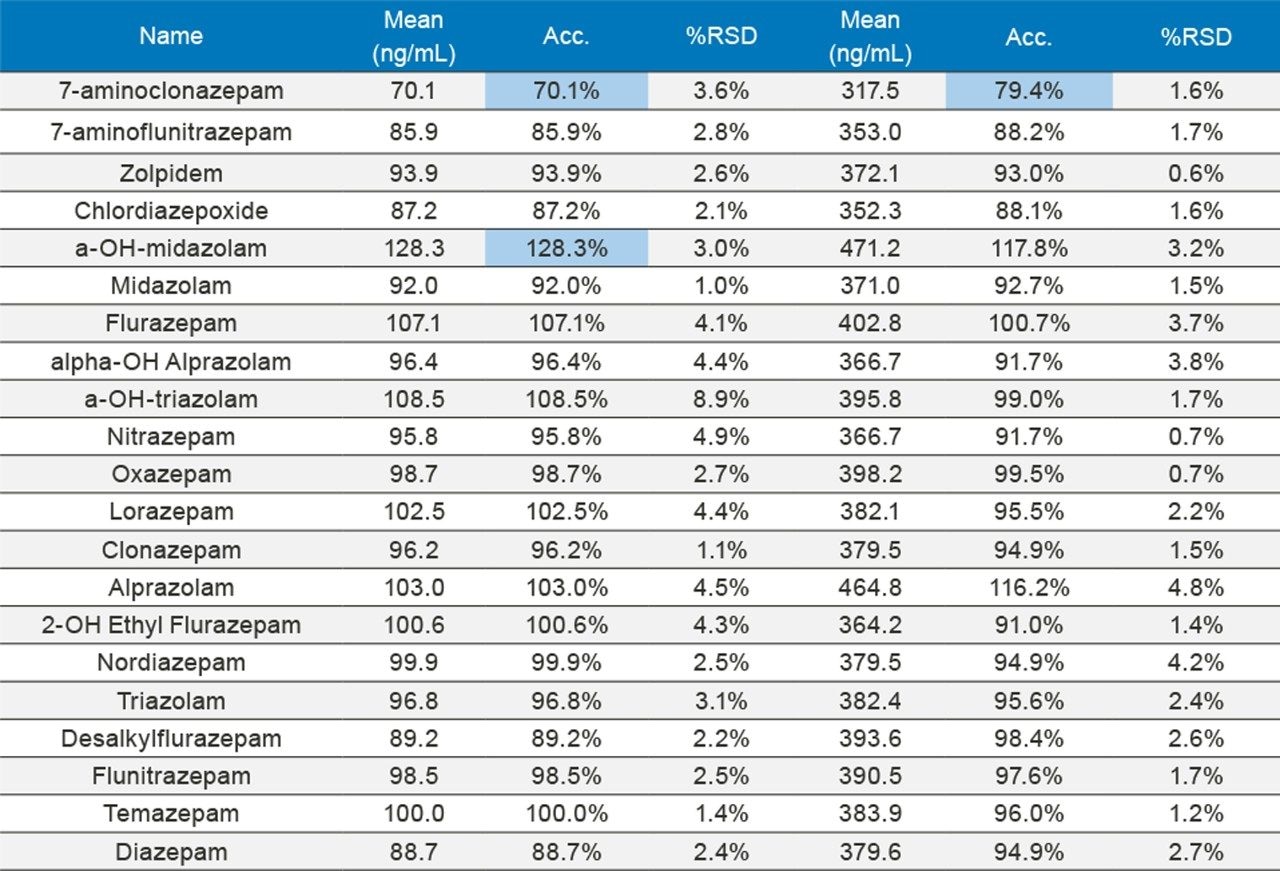
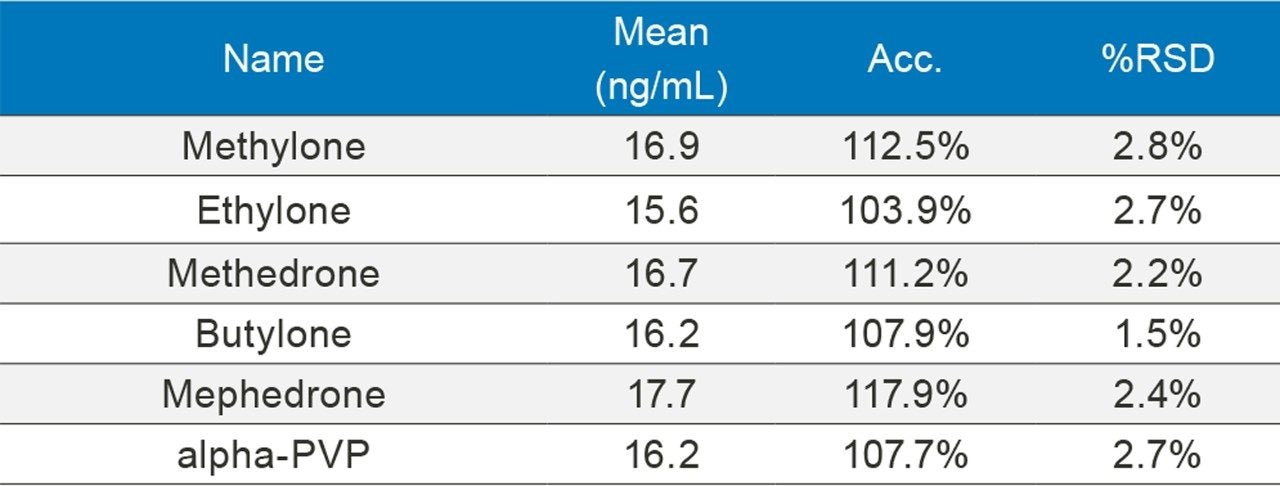
In order to assess accuracy, external quality control samples from UTAK Laboratories were evaluated. These results can be seen in Tables 2A–2D. Analytes assessed using external quality control samples included opioids, benzodiazepines, stimulants, and synthetic cathinones. These results show that 91/98 (93%) of the results were within 20% of the target value. The larger deviations for analytes such as fentanyl, norfentanyl, and buprenorphine could be a result of slight errors in the preparation of the master stock mix, as these compounds were spiked using low volumes (20 µL of stock solution). In addition, 7-aminoclonazepam may have stability issues in the urine matrix which could account for its low bias. All results had %RSD values <10%.