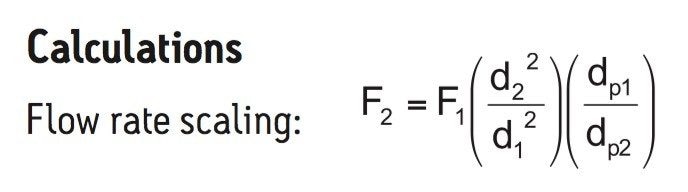ACQUITY UPLC H-Class Bio System for HPLC monosaccharide analysis
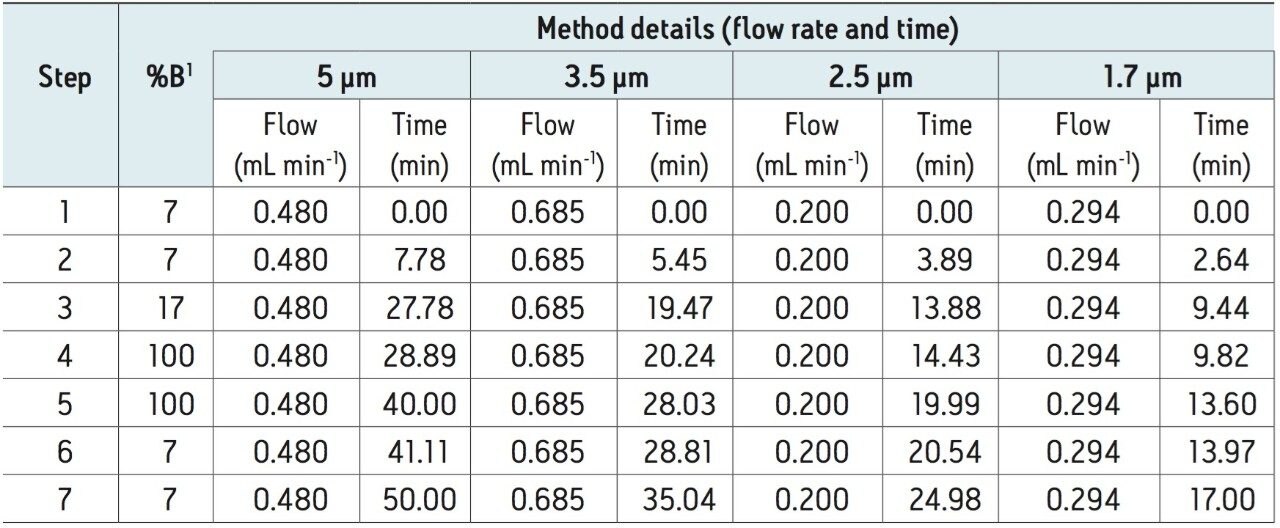
To verify the ability of the ACQUITY UPLC H-Class Bio to run legacy analyses of 2AA-derivatized monosaccharides, we first established an HPLC separation using method conditions based on previously described chromatographic conditions1,2 To evaluate the proposed method, a reference standard mix was prepared by combining individual monosaccharides into a common mix. This mix included the typical monosaccharides expected in biologically relevant samples, namely N-acetylglucosamine (GlcNAc), N-acetylgalactosamine (GalNAc), glucose (Glc), mannose (Man), galactose (Gal), xylose (Xyl), and fucose (Fuc). In addition to these standards, two glycoprotein samples were also selected to determine the accuracy of this approach in determining monosaccharide composition. The first glycoprotein selected was bovine fetuin, a protein known to contain both N- and O-glycosylation sites. The second glycoprotein selected was the commercial monoclonal antibody cetumixab.
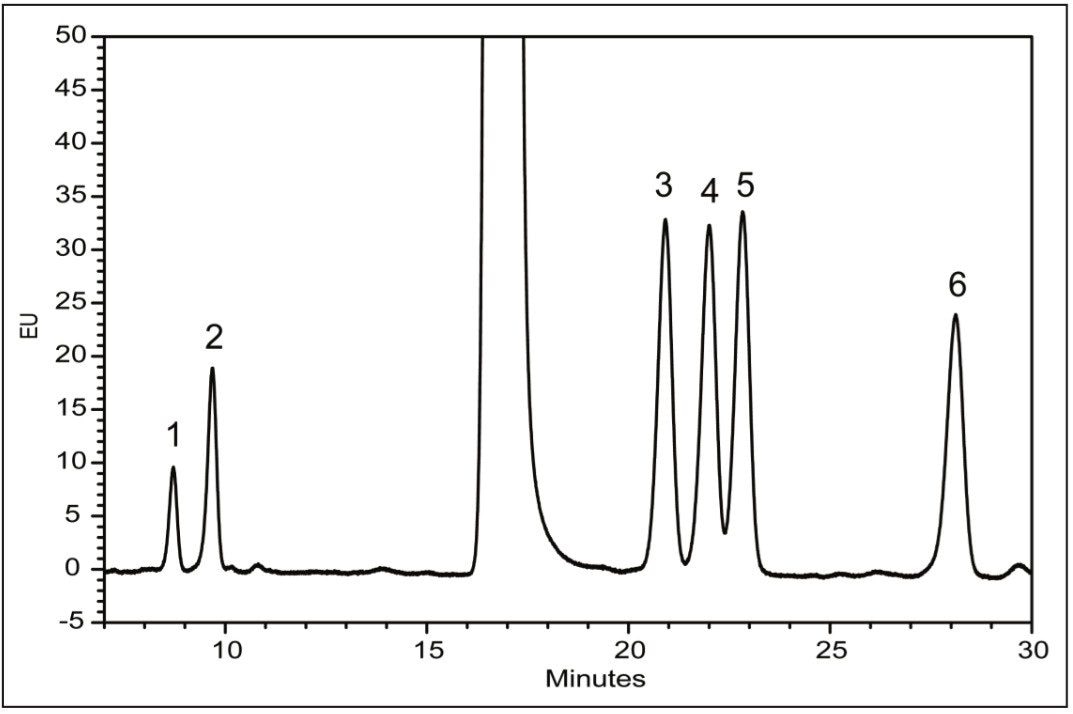
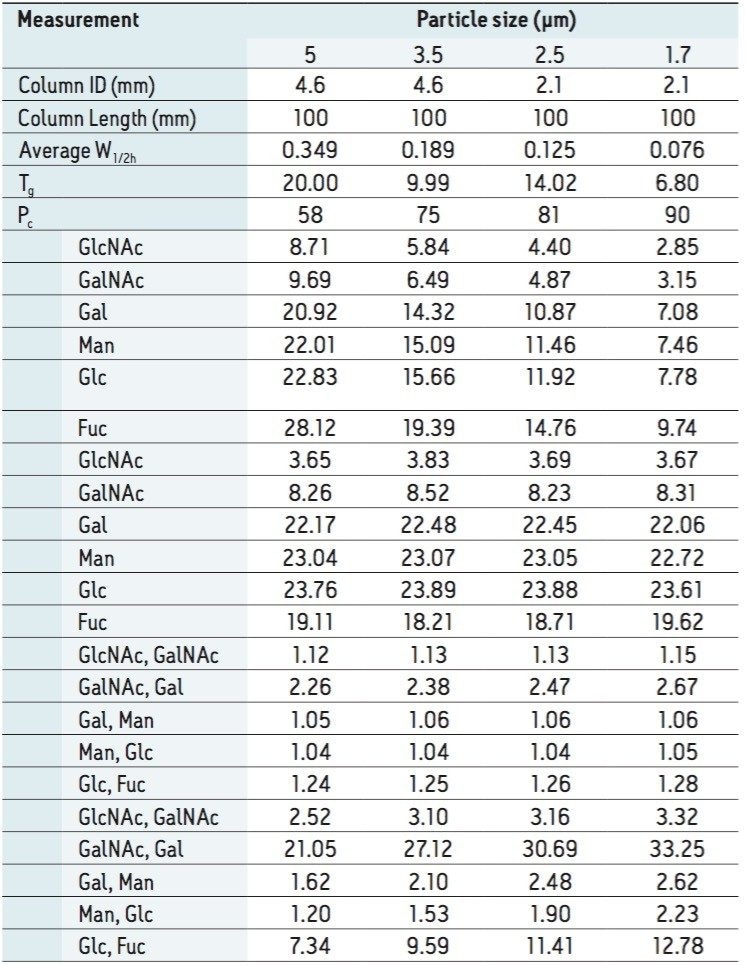
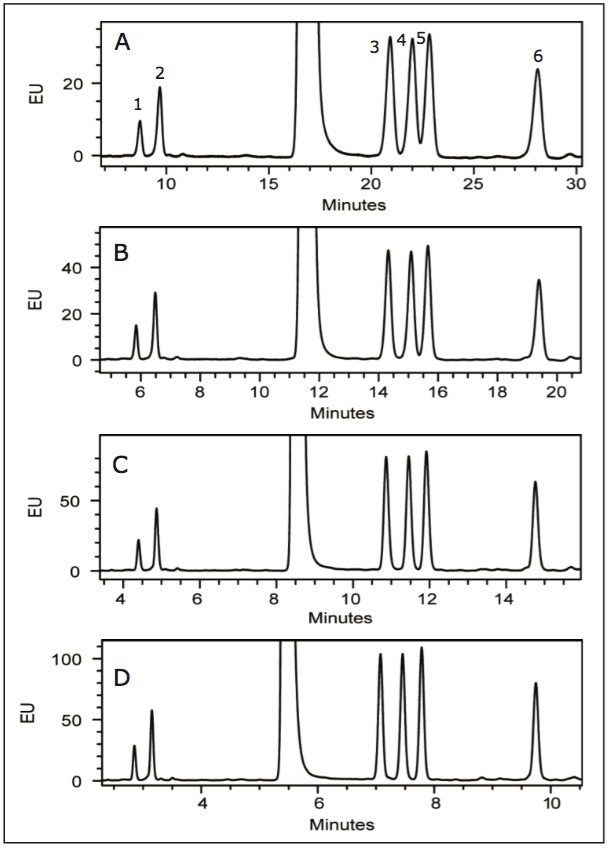
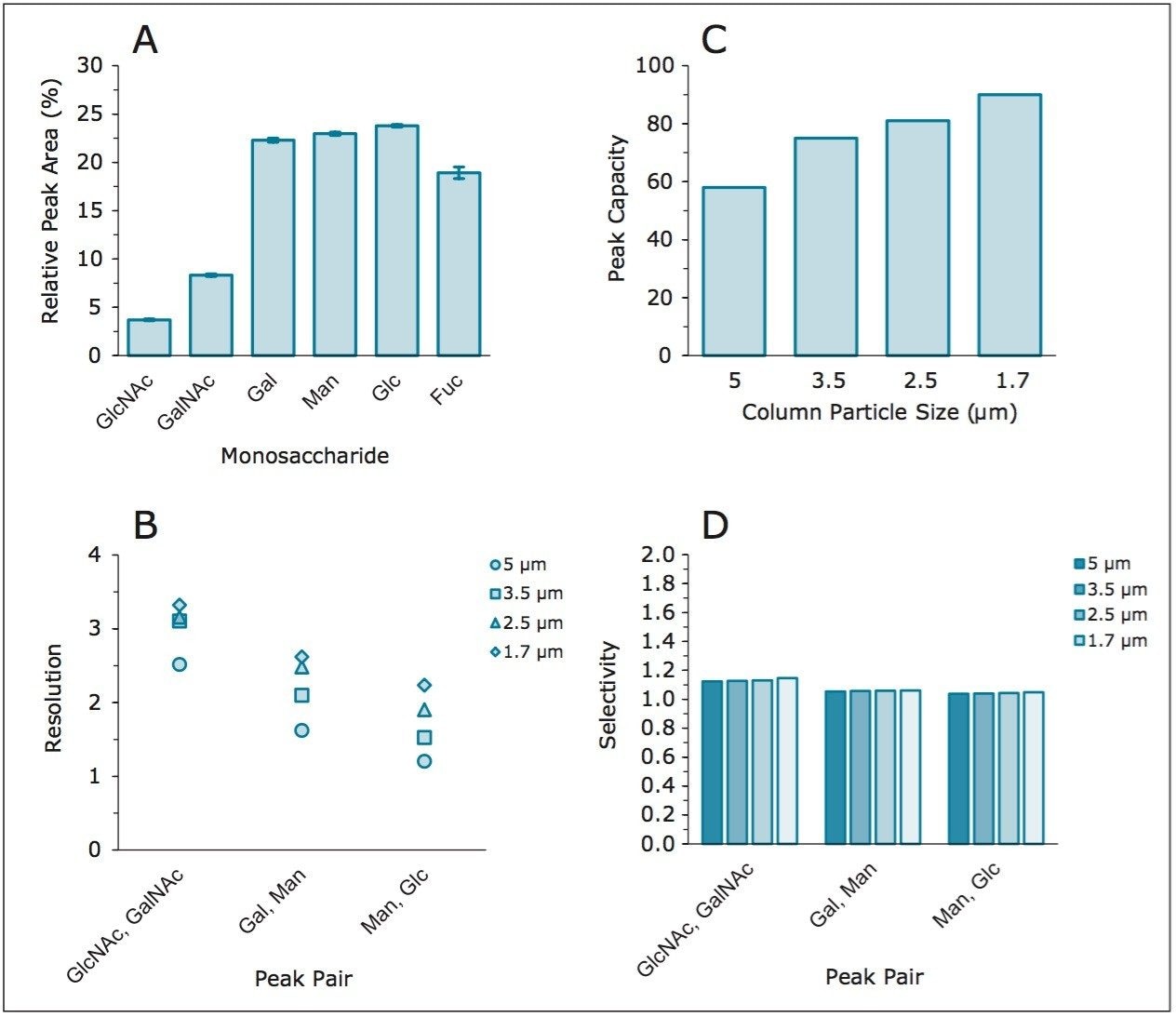
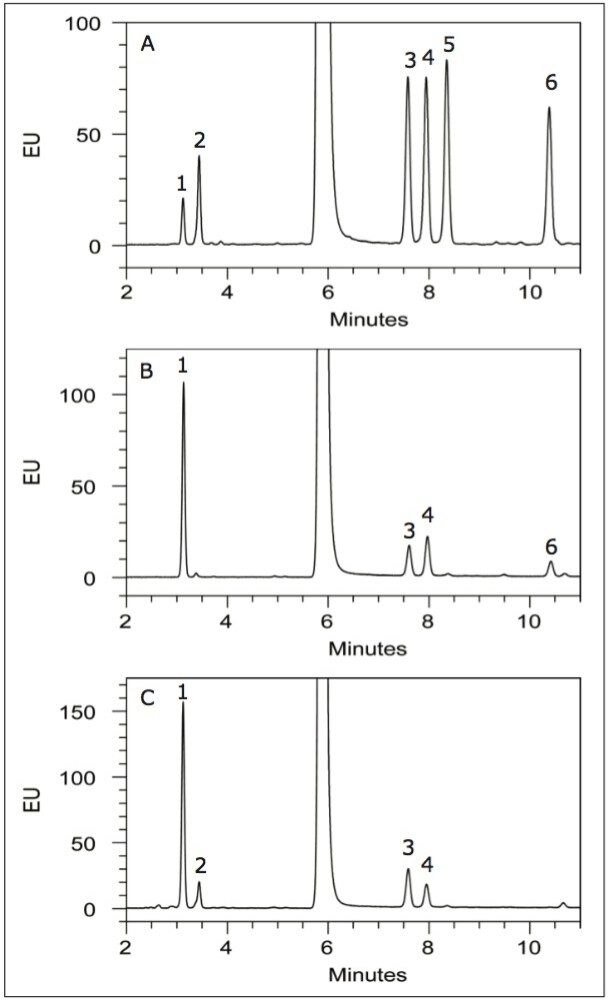
Following hydrolysis from the glycoprotein samples and 2AA derivatization, monosaccharides were separated using the aforementioned method. The resulting HPLC chromatogram (Figure 1) acquired on the ACQUITY UPLC H-Class Bio is consistent with previously published data.2 In terms of chromatographic performance, peak capacity was measured together with selectivity and resolution between critical peak pairs. These data are summarized in Table 1. Consistent peak area was observed across separations using all particle sizes (Figure 2d). These data confirmed the separation of all relevant components and, therefore, established a suitable method for monosaccharide method scaling to UPLC technology.