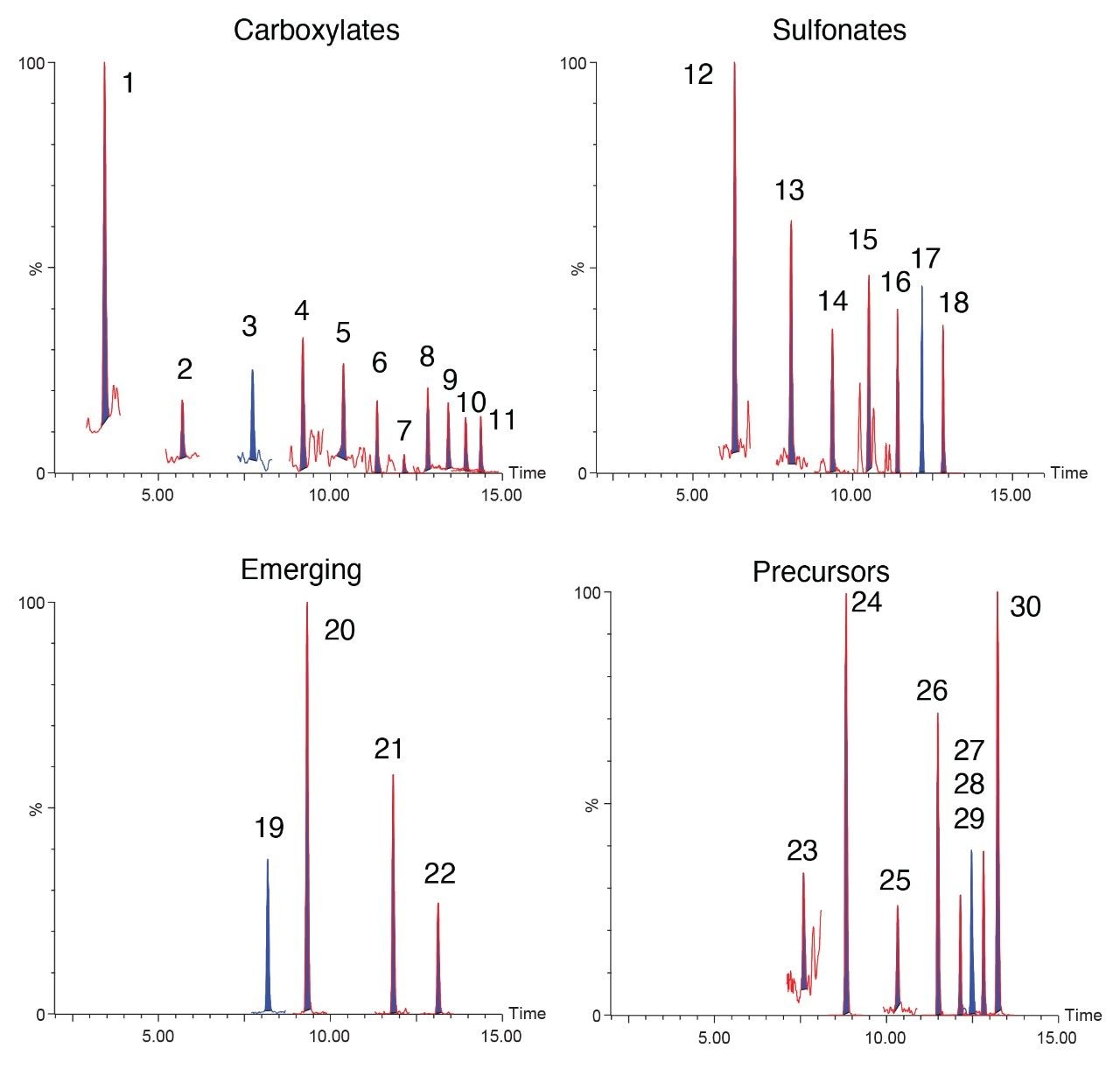
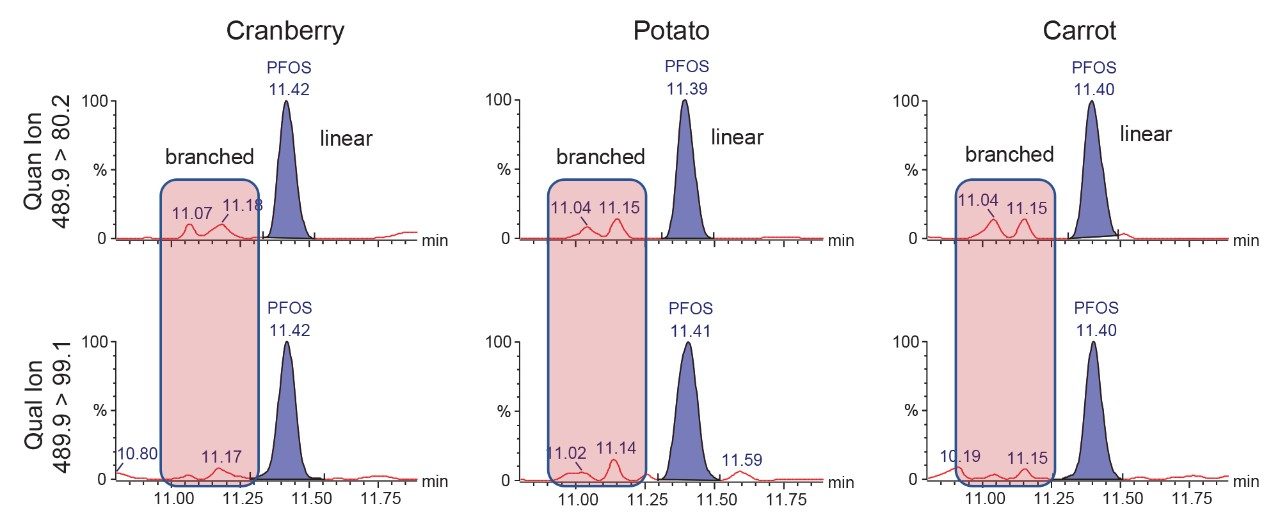
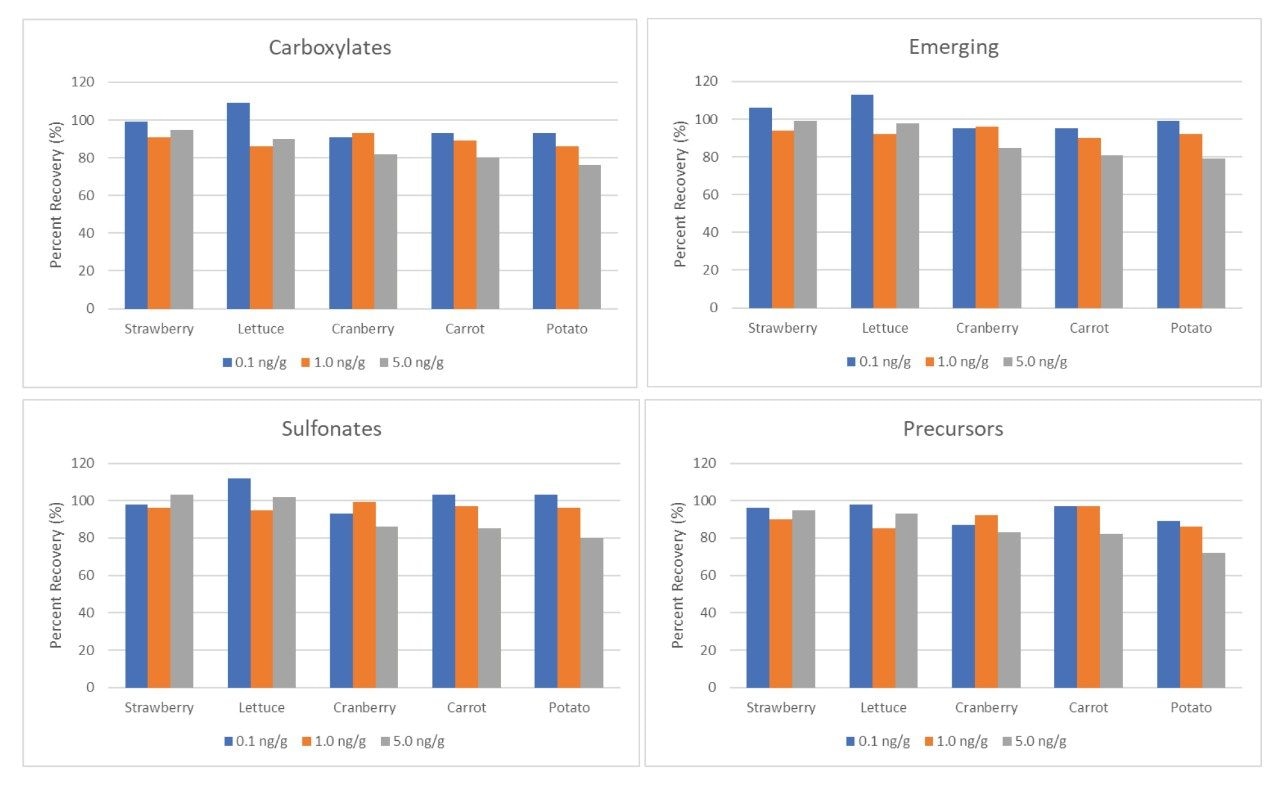
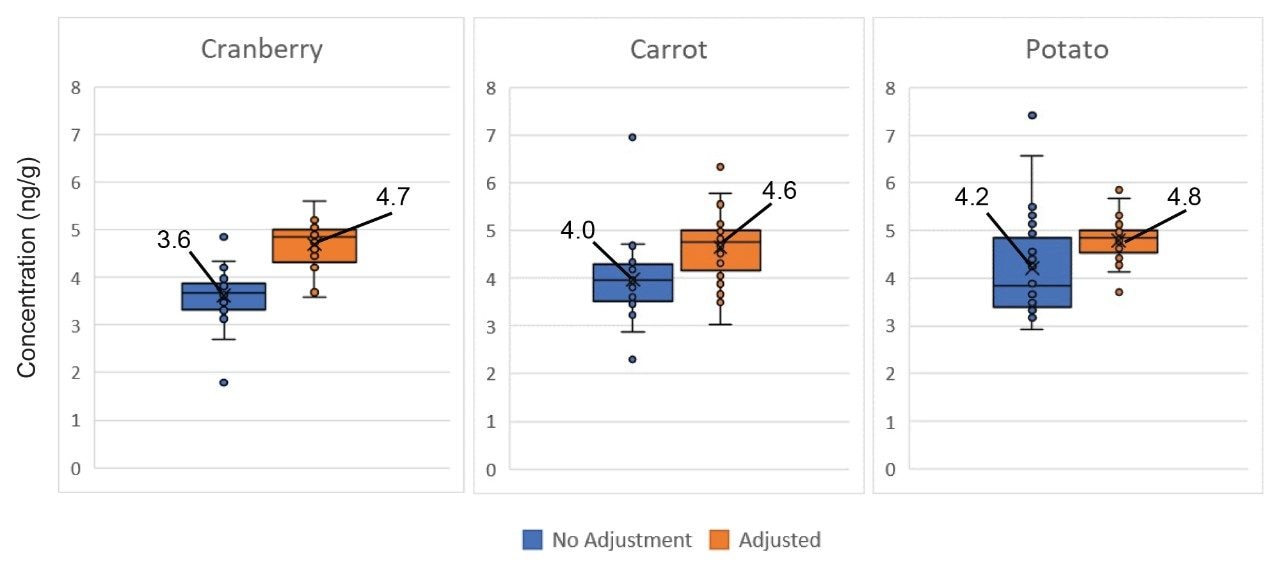
The environmental impact of per- and polyfluoroalkyl substances (PFAS) is readily known from the prevalent usage of these compounds in everyday products. Often, environmental issues also impact our food sources.
In the case of agricultural produce, PFAS impacted water and soil used for irrigation and growing crops can result in contamination. Studies show that edible plants do uptake PFAS, with higher uptake of short chain PFAS (PFBA and PFPeA) in the edible portions and a wider range of PFAS uptake in the roots and stalks/stems of the plants.1,2 Since irrigation water is most typically also drinking water or ground water, contamination can be introduced through any of the environmental contamination pathways (manufacturing discharge, firefighting foam, landfill leachate, etc).3 Soil contamination can occur from similar mechanisms, but the use of biosolids as fertilizers has become a major concern for crop contamination.4
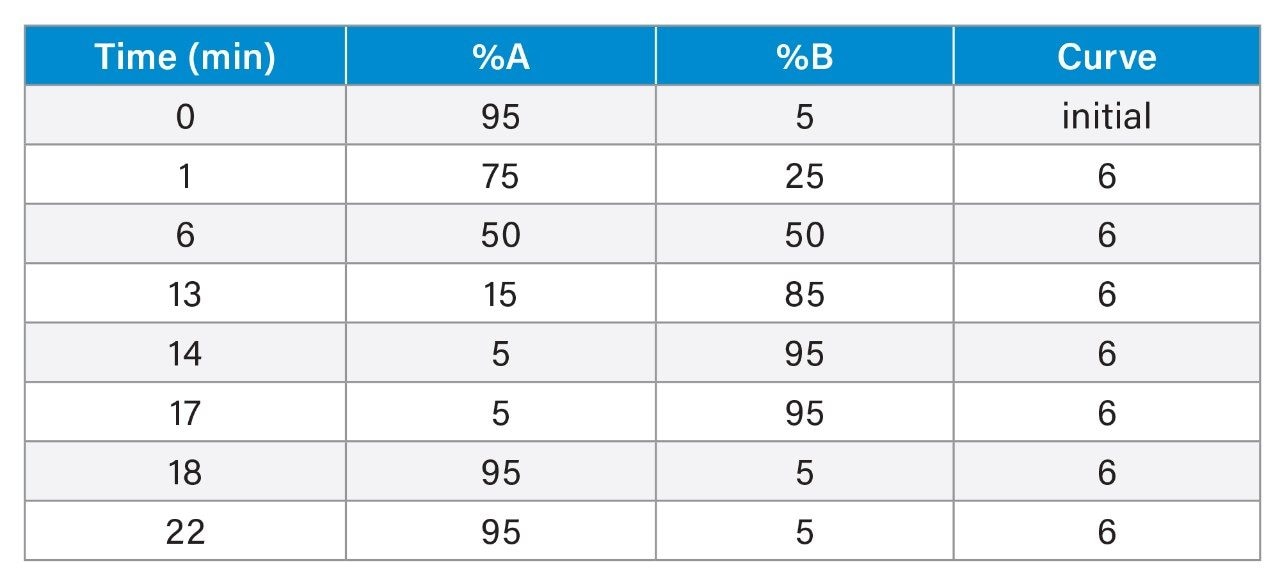
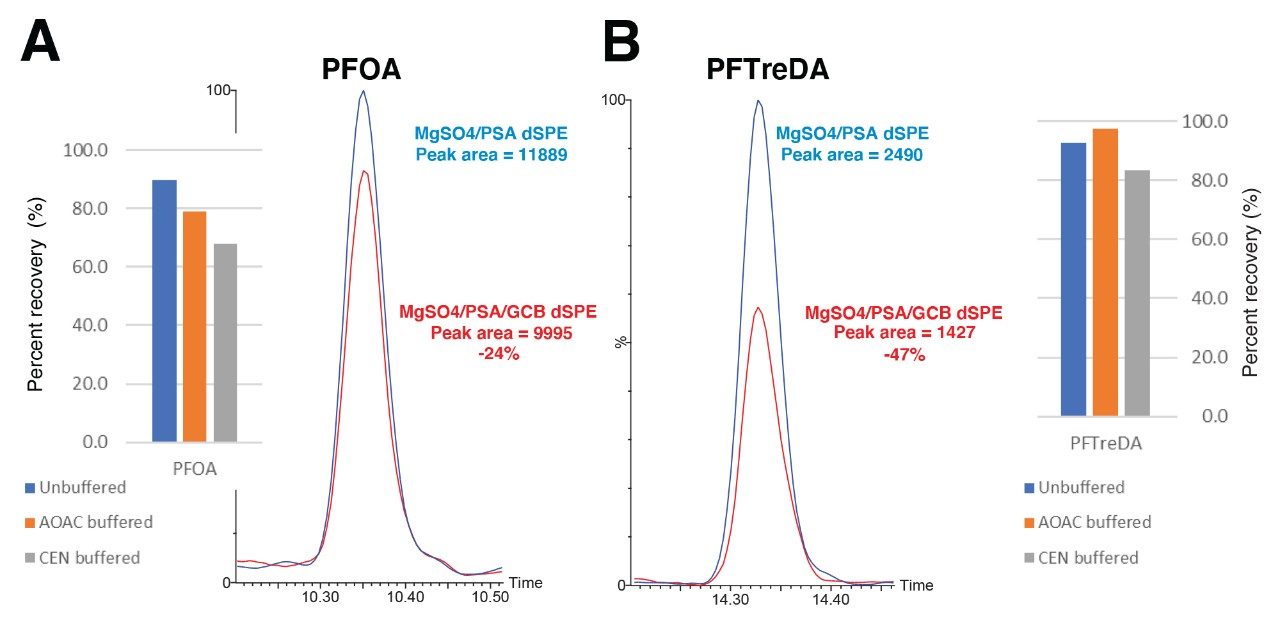
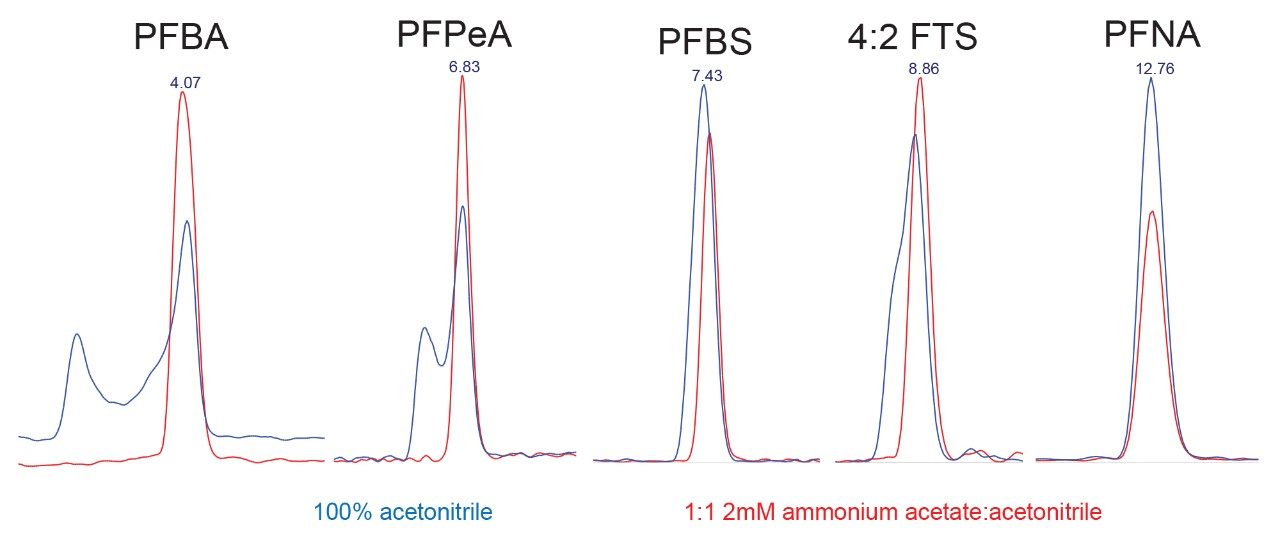
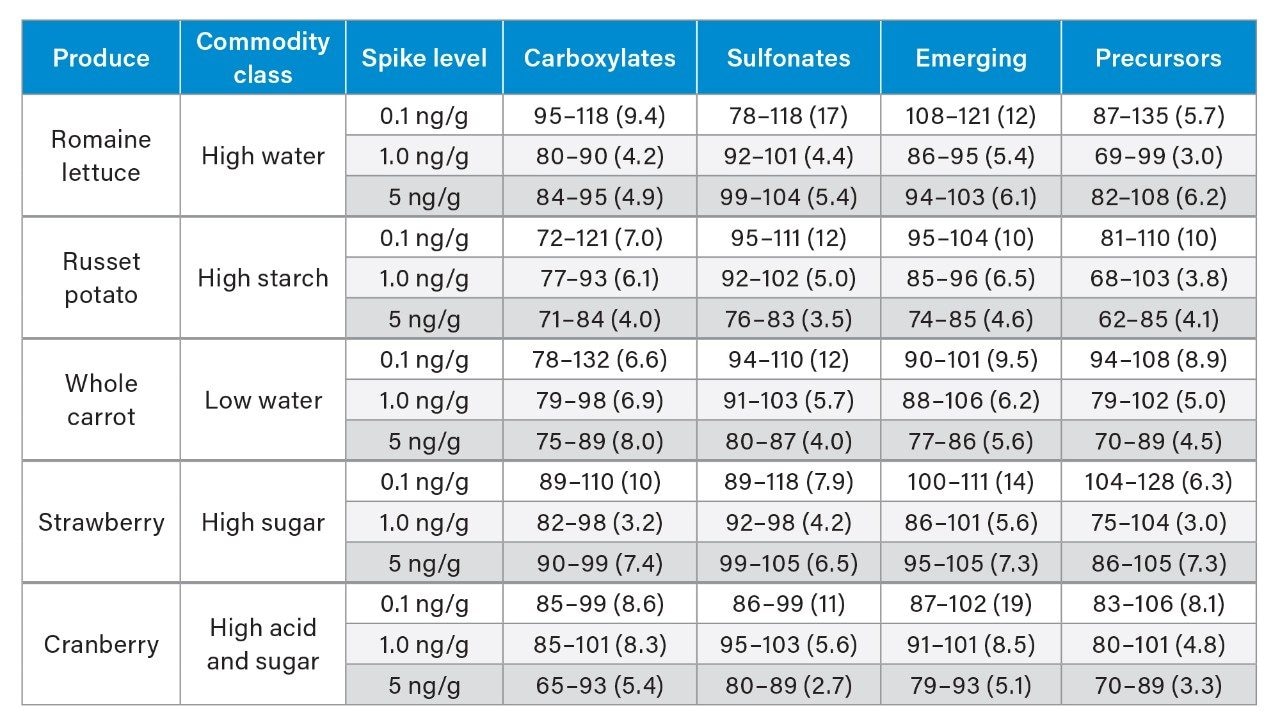
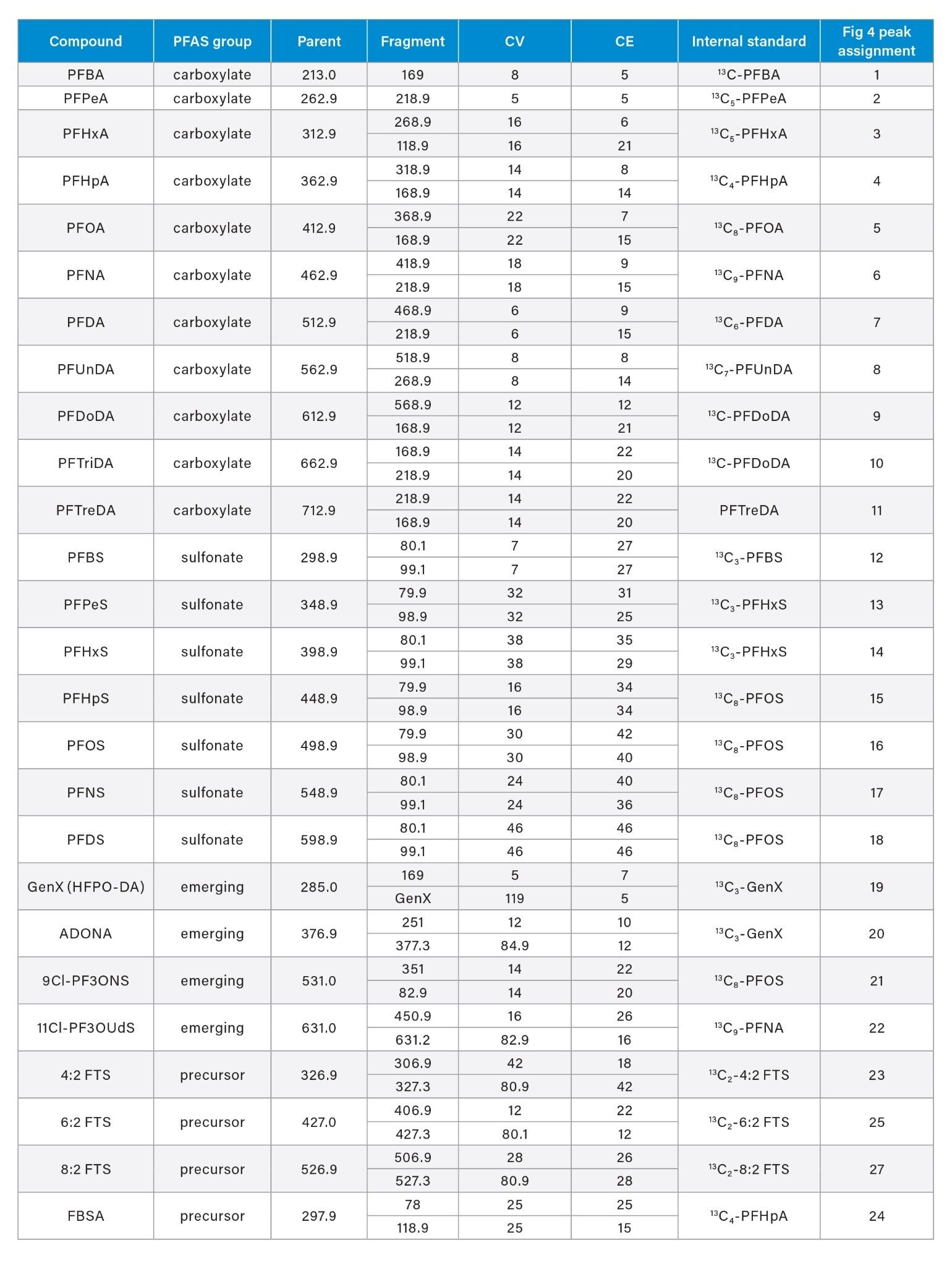
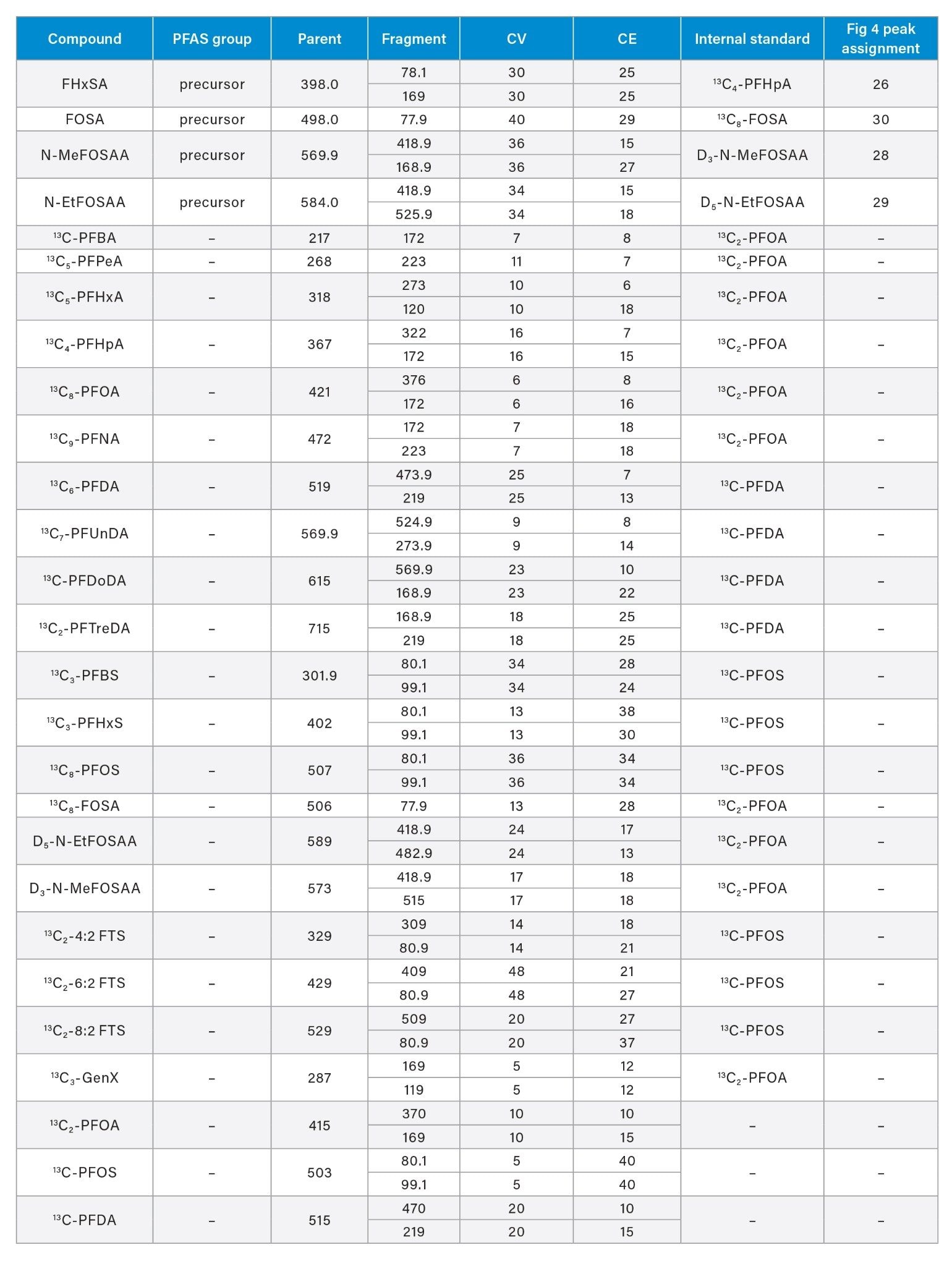
Although some countries impose regulatory or advisory limits on the concentration of PFAS in water, limits for PFAS have yet to be set in biosolids or food. The European Food Safety Authority (EFSA) have evaluated and published data on human health risks due to presence of PFAS in food. The most recent report published in 2020 concluded that, of the 27 PFAS evaluated, fish, fruit, and eggs contributed the highest levels of exposure.5 Data submitted for this study utilized a range of extraction and analysis techniques. The US Food and Drug Administration (FDA) monitor contaminants in highly consumed foods in their Total Diet Study.6 To be able to include PFAS in this study, they created and validated an extraction method for PFAS in foods.7 This method (FDA C-010.01)8 utilizes a QuEChERS extraction, followed by dispersive solid phase extraction (dSPE) clean up. QuEChERS (Quick, Easy, Cheap, Effective, Rugged, Safe) is a widely used extraction technique first created for extraction of pesticides from food and is often adopted for determination of other contaminants. This technique uses salts and acetonitrile to extract compounds of interest through a salting out and phase separation mechanism. This fast and simple extraction technique was evaluated for extraction of PFAS from a variety of produce samples, with analysis using ACQUITY UPLC I-Class PLUS coupled to Xevo TQ-XS.