Synthetic mRNA Oligo-Mapping Using Ion-Pairing Liquid Chromatography and Mass Spectrometry
Abstract
Messenger RNA has quickly become an important modality for human medicine, as shown with its evaluation for cancer treatment and the FDA approval of mRNA vaccines for COVID-19. The rapid development of mRNA vaccines and other classes of mRNA therapeutics is supported by advances in analytical methodologies. One important aspect of such methodologies is confirming the identity, purity, and modification(s) of a therapeutic mRNA through mapping its sequence by liquid chromatography-mass spectrometry (LC-MS). Mass spectrometry-based sequencing of RNAs has an advantage over templated RNA sequencing by offering direct molecular detection of fragments, which can be used to localize nucleoside impurities and identify important structural attributes (5’-cap and poly-A tail). As such, we propose a workflow for comprehensive bottom-up LC-UV-MS characterization of mRNAs that yields mRNA component-annotated chromatograms derived from accurate-mass matching.
Benefits
- High chromatographic resolution and MS sensitivity with the use of ion-pairing reversed-phase chromatography in combination with the ACQUITY™ Premier Oligonucleotide BEH™ C18 300 Å Column
- ·Automated mRNA digest annotation based on accurate-mass matching as facilitated by in silico mRNA digestion calculations and application of waters_connect/UNIFI™ Scientific Libraries
Introduction
The SARS-CoV-2 pandemic provided the impetus for the fast-paced development of nucleic-acid based medicine, especially synthetic mRNA.1 Now, 40 years after its discovery in 1961 by Brenner et al.2 [i], mRNA has evolved into being an important modality with massive potential, as shown with the start of an in-human clinical trial for cancer treatment1–3 and the full approval of two COVID-19 mRNA vaccines by the US Food and Drug Administration in August 2021 and in January 2022, respectively. The rapid development of mRNA vaccines and other classes of mRNA therapeutics are supported by advances in analytical methodologies. One important aspect of such methodologies is confirming the identity, purity, and modification of a therapeutic mRNA through oligo-mapping and sequencing via liquid chromatography hyphenated to mass spectrometry (LC-MS). Nucleic acid sequencing technologies, like Sanger and next-generation sequencing (NGS), provide valuable information to drug developers. However, there is also a heightened level of analysis that can be achieved through the use of LC in combination with tandem MS (LC-MS/MS)4 or MSE (alternating low and high collision energy)5 based fragmentation. Similar to a proteomics bottom-up approach, LC-MS/MS or MSE based sequencing have the advantage of direct molecular detection of RNA fragments, including the detection and localization of nucleoside impurities and important structural attributes, like lipidated nucleobases,6 endcapped residues, and polyA tail modifications.4,7 Unlike bottom-up proteomics workflows, where a plethora of data processing solutions exist, options for RNA mapping are limited. We propose a workflow for oligo-mapping based on a bottom-up approach for the characterization of a given synthetic mRNA within a single platform comprising LC, UV detection and MS measurements. Digestion components were processed using an in-house developed, freely available in silico digestion library calculator, mRNAcalcondemand, in combination with waters_connect™ to yield an annotated chromatogram. Here, we demonstrate this analytical approach for mRNA sequence mapping using RNase T1 digested luciferase mRNA.
Experimental
Sample Information
Approximately 90 µg of synthetic Cypridina luciferase mRNA (uncapped and not modified with a polyA tail); a gift from Bijoyita Roy (New England Biolabs, Ipswich, MA) was digested using 3’-guanosine specific ribonuclease RNaseT1 (Worthington Biochemical Corporation, Lakewood, NJ). Note that this workflow was repeated using 10 µg of TriLink Biotechnologies (CleanCap® FLuc mRNA, San Diego, CA) firefly luciferase mRNA (untranslated sequences are proprietary) and comparable results were achieved. Luciferase mRNA was denatured prior to digestion using 20 µL urea (8 M) prepared in nuclease-free buffer (10 mM Tris, 0.1 mM EDTA, pH 7.5 in water, Integrated DNA Technologies, Inc, Coralville, IA) at 80 °C for 5 minutes. Next, 24 µg (~10kU) of RNase T1 (Worthington Biochemical Corporation, Lakewood, NJ) resuspended in nuclease-free buffer was added to the denatured mRNA at room temperature and the mixture was then incubated at 37 °C for 30 minutes. Nuclease-free buffer (40 µL) was added at the end of the incubation period, bringing the total sample volume to 80 µL. The final aliquot was transferred to a polypropylene 300 µL autosampler vial (p/n: 186002639). The resulting digest was subjected to ion-pairing reversed-phase chromatography (IP-RPLC) without any further manipulation prior to MS detection in negative ion mode using the BioAccord™ RDa™ Detector.
LC Conditions
|
LC system: |
ACQUITY UPLC™ Premier BSM System (as part of the BioAccord System) |
|
Detector: |
ACQUITY UPLC TUV Detector |
|
Wavelength: |
260 nm |
|
Column: |
ACQUITY Premier Oligonucleotide BEH C18, 2.1 X 150 mm, 300 Å, 1.7 µm (p/n:186010541) |
|
Column Temperature: |
70 ˚C |
|
Sample Temperature: |
4 ˚C |
|
Injection: |
5 μL |
|
Flow Rate: |
0.4 mL/min |
|
Mobile phase A |
0.1% N,N-diisopropylethylamine (DIPEA) as the IP reagent and 1% 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) in deionized water |
|
Mobile phase B |
0.0375% DIPEA and 0.075% HFIP in 65:35 acetonitrile/water |
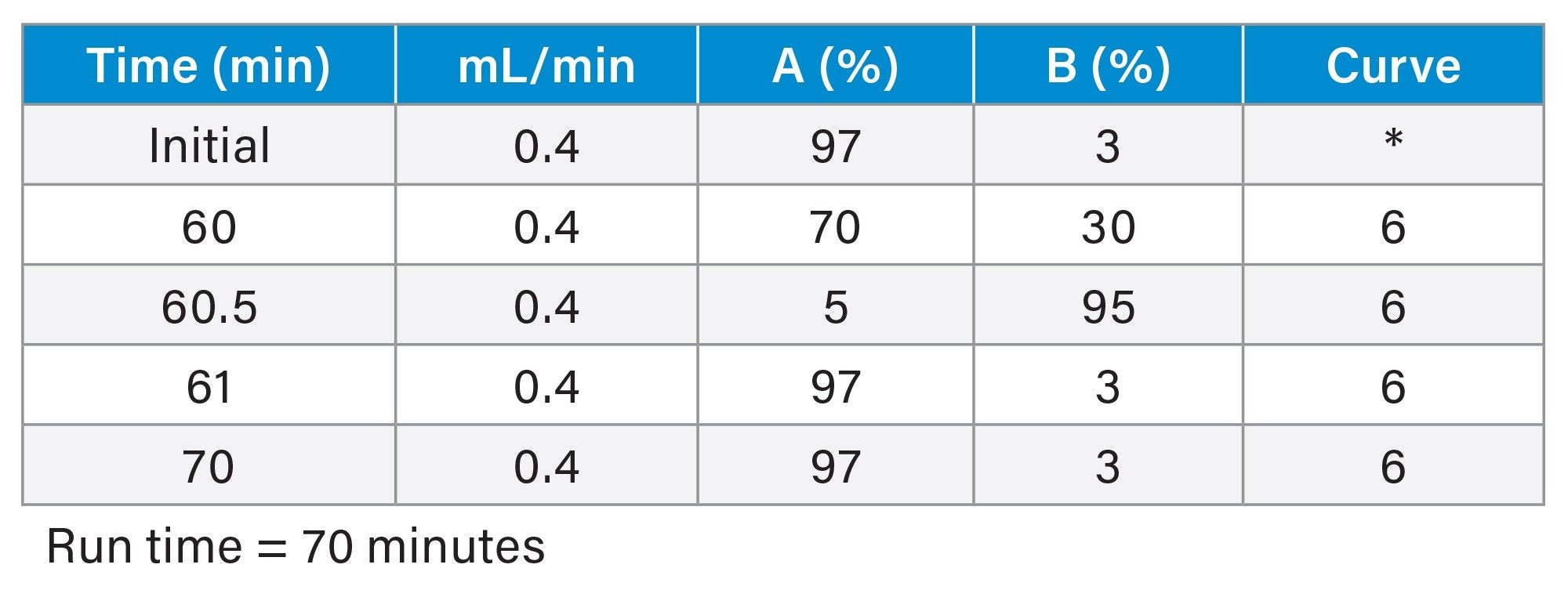
Gradient Table
MS Conditions
|
MS system: |
BioAccord LC-MS System |
|
Detector: |
ACQUITY RDa Detector |
|
Mode: |
Full scan with fragmentation |
|
Polarity: |
Negative |
|
Cone voltage: |
40 V |
|
Fragmentation cone voltage: |
80–200 V |
|
Mass range: |
High (400–5000 m/z) |
|
Scan rate: |
2 Hz |
|
Capillary voltage: |
0.80 kV |
|
Desolvation temperature: |
400 °C |
Results and Discussion
Ion-pairing reversed-phase chromatography (IP-RPLC) with a C18 stationary phase has become a tried-and-true approach for the analysis of oligonucleotides.4,7 The mobile phase contains an ion pairing reagent, commonly an alkylamine, which adsorbs onto the C18 stationary phase8,9,10 and thereby introduces a mixed mode like retention mechanism.8–10 The N,N-diisopropylethylamine (DIPEA)/ 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) mobile phase system used in this application is compatible with both optical UV detection and negative ion mode mass spectrometry.4,7,–10 HFIP is used to enhance electrospray ionization.8
RNase T1 digested luciferase mRNA was injected onto an ACQUITY Premier Oligonucleotide BEH C18 (2.1 x 150 mm, 300 Å, 1.7 µm) Column and gradients were developed with an ACQUITY Premier Binary LC equipped with an ACQUITY UPLC TUV Detector. The ACQUITY Premier Oligonucleotide BEH C18 Column used in this work is similar to ACQUITY Premier Oligonucleotide BEH C18 130 Å Column, but with a wider pore size, providing better resolution for longer oligonucleotide species. Data were acquired in triplicate using negative ion mode mass spectrometry with the ACQUITY RDa Detector of a BioAccord Benchtop LC-MS system. Moreover, the ACQUITY RDa Detector was programmed to acquire MSE data such that every other scan produced a high energy fragment ion spectrum that could be later used to corroborate LC peak identifications. Figure 1 depicts total ion chromatograms (TIC) for an RNase T1 control sample (top trace), mRNA control sample (bottom trace) and the digested mRNA (middle trace). Oligonucleotide fragments resulting from the digestion of luciferase mRNA with RNase T1 were readily separated according to a separation with a 4-sigma peak capacity of 613. Overall, chromatographic peaks were sharp and symmetrical with ~0.01 minutes variation in retention time (RT) over triplicate injections. Digest components eluted between 2 and 23 minutes on a 60 minute gradient time method; some incompletely digested mRNA was seen to elute around 29 minutes and intact RNaseT1 eluted around 54 minutes. As can be seen on the top trace of Figure 1, an RNase T1 control sample showed signal only after a 50 minute retention time. This confirmed that RNase T1 would not introduce interference within the retention window of the mRNA digestion components. (Figure 1, middle trace). Likewise, the bottom trace of Figure 1 shows that intact luciferase mRNA elutes at approximately 38 minutes, confirming that the peak observed at 29 minutes in the digested sample (Figure 1, middle trace) corresponds to incompletely digested mRNA. We note here that in the case of synthetic mRNA comprising 5’-cap and poly-A tail structures, peaks at ~29 and 37 minutes are observed post digestion. The slight shift from ~38 minutes to ~37 minutes might be indicative of the undigested polyA structure (an investigation into this chromatographic behavior will be described in a future application note).
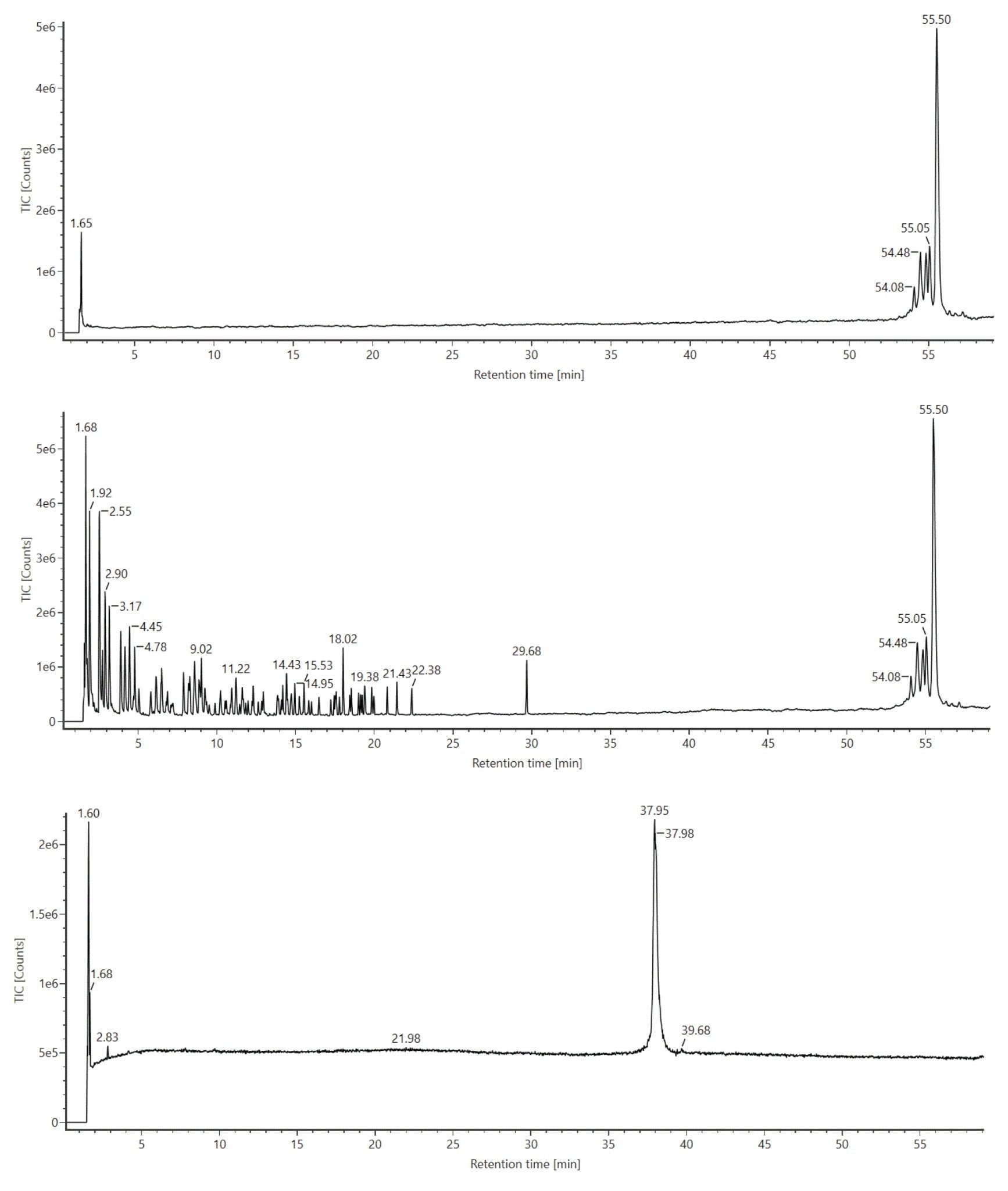 Figure 1: TIC for RNase T1 control sample (top trace), mRNA control sample (bottom trace) and the digest (middle trace) obtained from ion-pairing reversed-phase chromatography (IP-RPLC) of luciferase mRNA digested with Rnase T1 and analyzed using the ACQUITY UPLC I-Class System (ACQUITY Premier Oligonucleotide BEH C18 Column, 2.1 x 150 mm, 300 Å, 1.7 µm) and the BioAccord ACQUITY RDa Detector in negative ion mode.
Figure 1: TIC for RNase T1 control sample (top trace), mRNA control sample (bottom trace) and the digest (middle trace) obtained from ion-pairing reversed-phase chromatography (IP-RPLC) of luciferase mRNA digested with Rnase T1 and analyzed using the ACQUITY UPLC I-Class System (ACQUITY Premier Oligonucleotide BEH C18 Column, 2.1 x 150 mm, 300 Å, 1.7 µm) and the BioAccord ACQUITY RDa Detector in negative ion mode.
The graphical user interface (GUI) of the in-silico digestion mRNA calculator, mRNAcalcondemand, is shown in Scheme 1. Next to the base sequence, a number of digestion parameters are specified, such as modification(s), enzyme, and missed cleavages. Based on this input, the calculator defaults to a number of MS specific settings, including charge state and m/z ranges, as well as the ability to conduct calculations based on monoisotopic or average mass. The generated output, in the form of a flat text csv file, can be utilized in UNIFI or waters_connect software, or used for complementary downstream analysis.
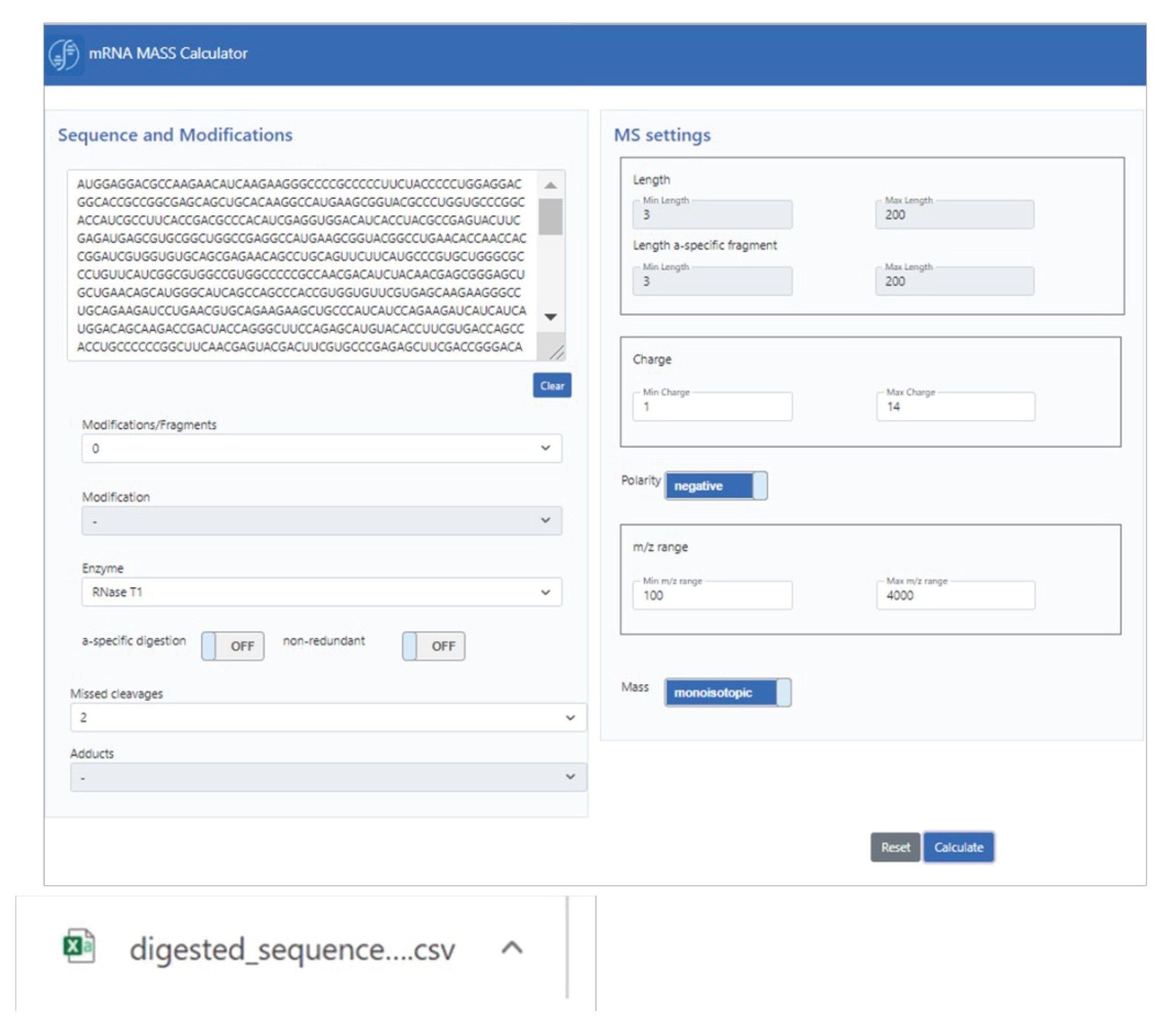 Scheme 1: mRNA calculator GUI for in silico mRNA digest mass calculation.
Scheme 1: mRNA calculator GUI for in silico mRNA digest mass calculation.
Next, a library was created where the digest components are considered to be individual analytes, which was achieved by importing the output of the calculator as a spreadsheet into a UNIFI Scientific Library. The created libraries can be utilized in an HRMS Screening Analysis Method, targeting the digest compounds post-acquisition, using user-specified tolerances. The use of mass tolerance-based library matching is demonstrated below. As a result of the library search, an annotated chromatogram is automatically generated (Figure 2). A close-up, corresponding to a zoomed view of the 17 to 20 minute retention time window is represented in Figure 2.
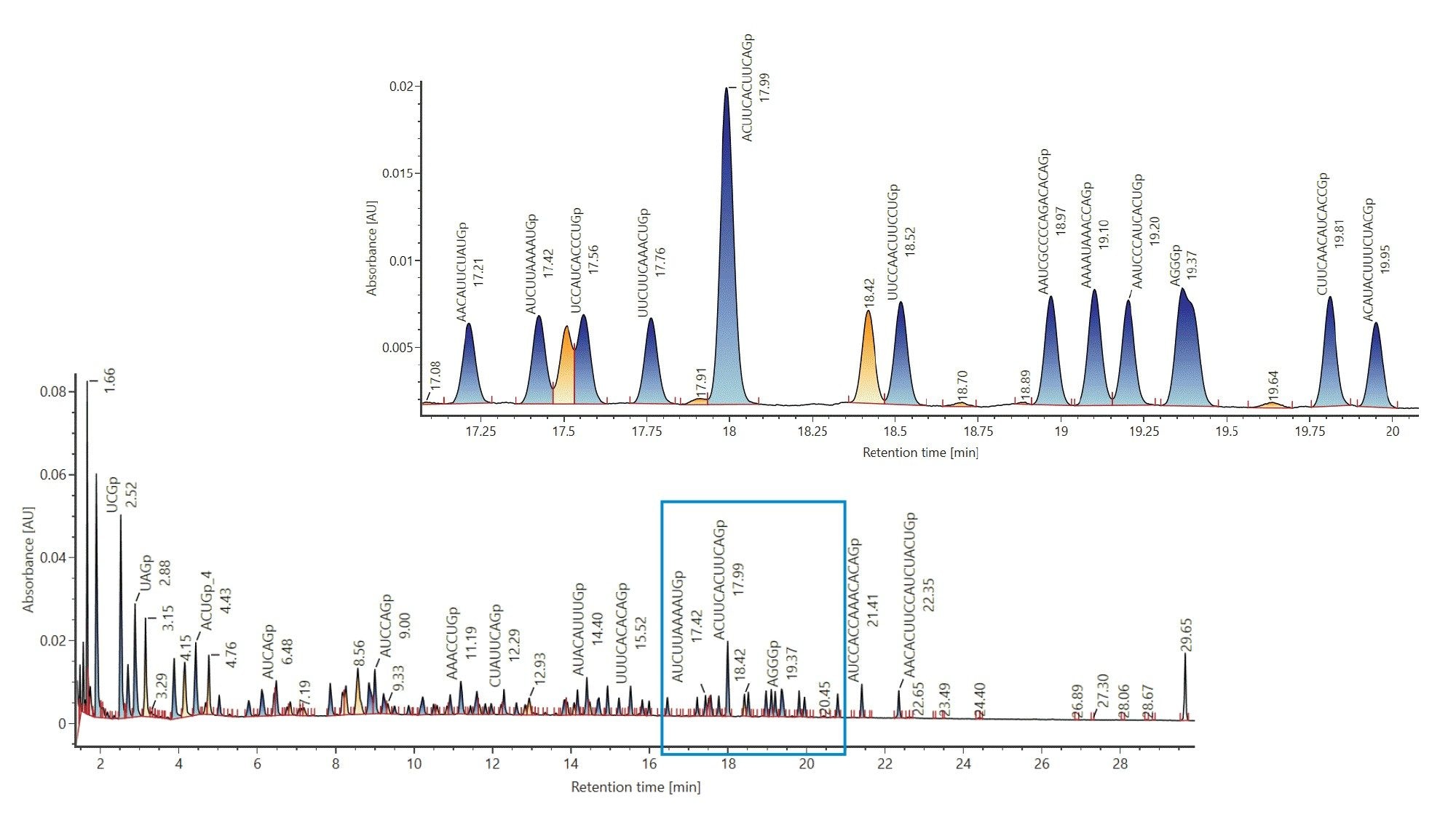 Figure 2: Annotated TIC of luciferase mRNA digest generated after matching, based on accurate mass, to a target component library. Luciferase mRNA was digested with Rnase T1 and analyzed using the ACQUITY Premier BSM LC (ACQUITY Premier Oligonucleotide BEH C18 Column, 2.1 x 150 mm, 300 Å, 1.7 µm) and the BioAccord ACQUITY RDa Detector in negative mode. Target components were calculated using the mRNA MASS calculator.
Figure 2: Annotated TIC of luciferase mRNA digest generated after matching, based on accurate mass, to a target component library. Luciferase mRNA was digested with Rnase T1 and analyzed using the ACQUITY Premier BSM LC (ACQUITY Premier Oligonucleotide BEH C18 Column, 2.1 x 150 mm, 300 Å, 1.7 µm) and the BioAccord ACQUITY RDa Detector in negative mode. Target components were calculated using the mRNA MASS calculator.
A total of 436, 428, and 441 potential identifications (IDs) of digestion components were produced from each technical replicate upon screening data against an in silico library created with an allowance for up to 2 missed cleavages. Several criteria were considered for manual validation. 40 out of 441 identified components were rejected based on abundance and peak shape, e.g., low abundant chromatographic peak shoulders. The majority of these rejected IDs (27 out of 40) were situated between 24 and 60 minutes. Overall, ~60% of the identified and validated components were within 10 ppm mass error (261 digest components). The RNase T1 control sample (Figure 1, top trace) was subjected to the same query and as expected did not yield any identifications. In addition to peak shape and abundance, we further validated the results based on high-confidence interpretation of isotopic distributions to deduce charge assignments, which yielded 139 digest components (65% within 5 ppm mass error). Lastly, some assignments were not included in the analysis because they appeared to be redundant assignments triggered by repeated detection on broadened and shoulder-containing chromatographic peaks. The number of identifications was thereby reduced by another 16 components. In a couple instances, however, analyte masses showed up at multiple retention times to produce two unique IDs corresponding to two distinct, well-defined chromatographic peaks. Here, we also took note of the presence of isomeric IDs (species having the same chemical composition but different sequences) as well as isobaric IDs (species having different chemical compositions [different exact mass], yet similar nominal mass). Ultimately, 90 unique components could be identified based on accurate-mass matching, and these are reported in Table 1, including isomeric nucleotide sequences and isobaric ions (cells highlighted in grey in Table 1).
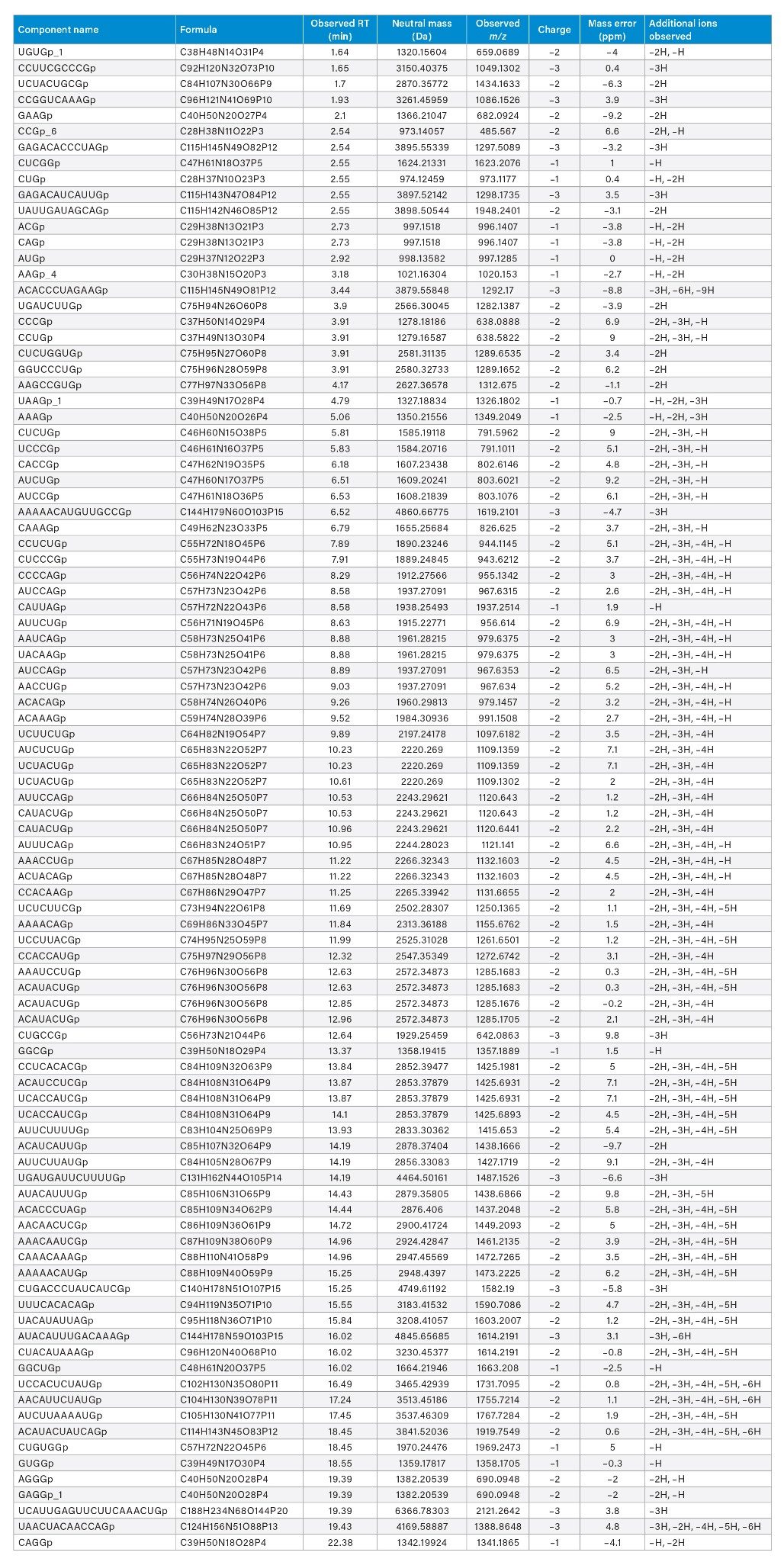 Table 1: Tentatively identified and validated luciferase mRNA digest components within a 10 ppm mass error based on accurate-mass matching. Cells corresponding to isomeric sequences or isobaric ions are highlighted in grey.
Table 1: Tentatively identified and validated luciferase mRNA digest components within a 10 ppm mass error based on accurate-mass matching. Cells corresponding to isomeric sequences or isobaric ions are highlighted in grey.
Figure 3 shows example data that produced an identification of digestion component UCCACUCUAUGp. This component eluted at 16.49 minutes as seen on the top left chromatogram and was identified from the in silico digestion library based on 5 ions carrying 2 to 6 negative charges (Figure 3, left bottom trace) at m/z 576.5732 ([M-6H]6-), 692.0876 ([M-5H]5-), 865.3603 ([M-4H]4-), 1154.1438 ([M-3H]3-) and 1731.7095 ([M-2H]2-). Isotopic distributions for [M-5H]5- and [M-2H]2- ions are illustrated to show data in support of charge state assignments (Figure 3, right).
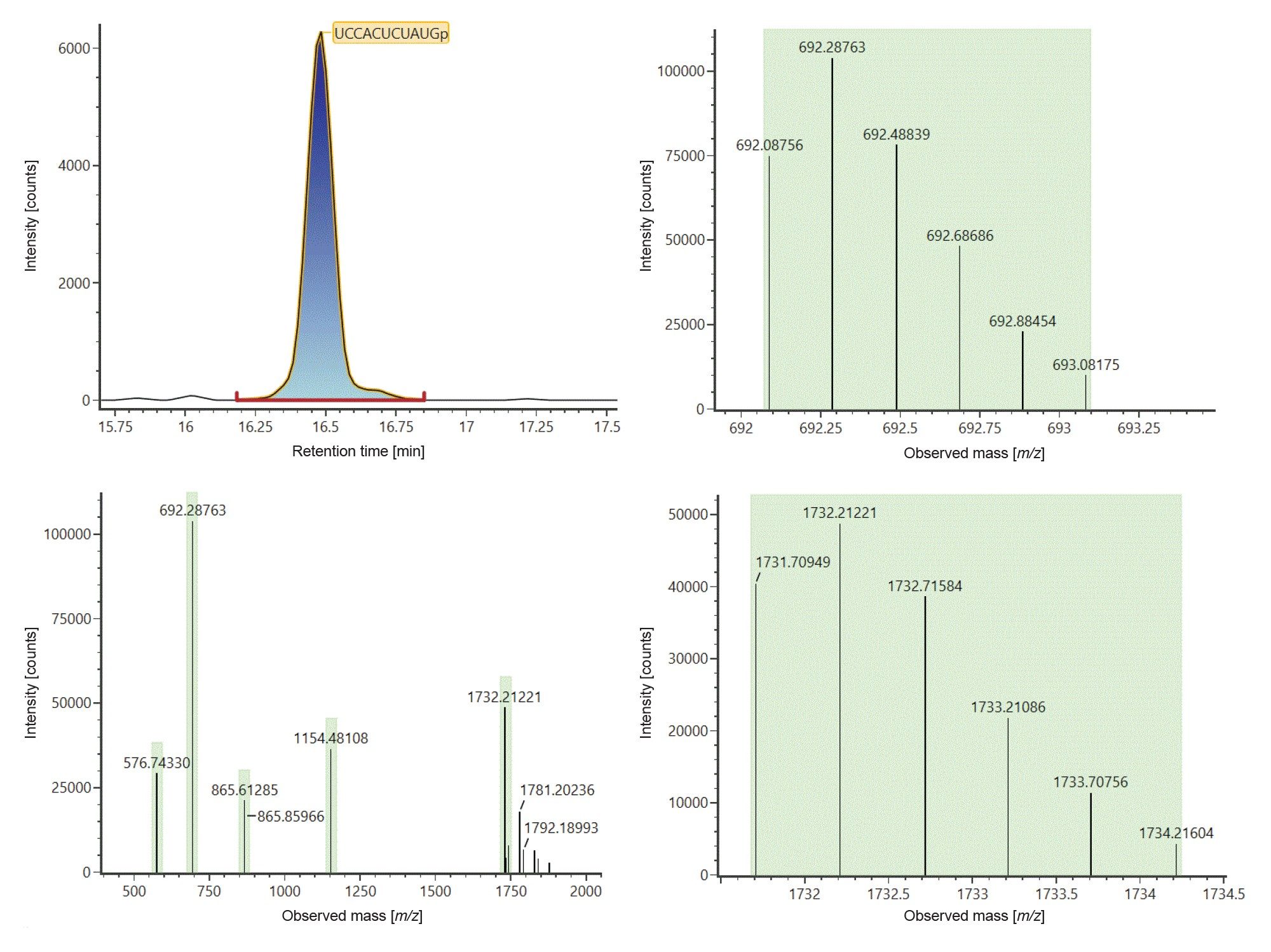 Figure 3: Identification of digest component UCCAUCACCCUGp eluting at 16.49 minutes (left top trace). The component was identified from the generated in silico digest based on 5 ions carrying 2 to 6 negative charges (left bottom trace) at m/z 576.5732 ([M-6H]6-), 692.0876 ([M-5H]5-), 865.3603 ([M-4H]4-), 1154.1438 ([M-3H]3-) and 1731.7095 ([M-2H]2-). The experimentally observed isotopic distributions for [M-5H]5- and [M-2H]2- ions are depicted on the right top and bottom traces respectively.
Figure 3: Identification of digest component UCCAUCACCCUGp eluting at 16.49 minutes (left top trace). The component was identified from the generated in silico digest based on 5 ions carrying 2 to 6 negative charges (left bottom trace) at m/z 576.5732 ([M-6H]6-), 692.0876 ([M-5H]5-), 865.3603 ([M-4H]4-), 1154.1438 ([M-3H]3-) and 1731.7095 ([M-2H]2-). The experimentally observed isotopic distributions for [M-5H]5- and [M-2H]2- ions are depicted on the right top and bottom traces respectively.
Ambiguities resulting from the presence of isomeric or isobaric ions were resolved using the waters_connect CONFIRM Sequence™ application for the interpretation of MSE spectra as reported in Table 2. Isomeric sequences at position 623–631 (ACAUCCUCGp) and 551–559 (UCACCAUCGp) are predicted from the RNAse T1 in silico digestion and both are assigned to the same retention time of 13.87 minutes. The correct assignment cannot be made using the intact mass analysis. MSE data from the same injection were used to elucidate the correct sequence for this assignment using the waters_connect™ CONFIRM Sequence application, wherein high energy fragment ions are predicted for each sequence and matched to isotope clusters of the integrated raw data via a bespoke algorithm. Confirmed fragment ions are represented on a Dot-Map allowing a rapid assessment of the sequence coverage (Figure 4, B): full sequence coverage was obtained for UCACCAUCGp such that it could be readily validated as the correct assignment (Table 2, index #63).
Neutral precursor masses obtained for manually validated digestion components (Table 1) spanned from 973.1406 Da (CCGp, RT 2.54 minutes) to a 20-mer nucleotide at 6366.7830 Da (UCAUUGAGUUCUUCAAACUGp, RT 19.39 minutes). The earliest eluting component was UGUGp, and it was identified with an observed neutral mass of 1320.1560 and a retention time of 1.64 minutes. The last luciferase mRNA digest component to elute within the manually validated set of IDs (Table 1) was CAGGp (1342.1992Da), and it was observed to elute at 22.38 minutes. Elution of CAGGp at such a late retention time was unexpected since the closely related sequence UAAGp eluted at ~4 minutes. In order to address the issue of false positives, we used the waters_connect CONFIRM Sequence application to further characterize components IDs. Validated based on MS data with MSE data. As it can been seen in Table 2, 34/90 components, including CAGGp, did not generate an adequate number of MSE fragments to further validate the sequence assignment, although accurate-mass matching supported the assignment. Another interesting example is the observation within the same dataset of assigned sequences AAAAACAUGUUGCCGp (4860.6678 Da, 15-mer, 9 purines) and AUACAUUUGACAAAGp (4845.6568 Da, 15-mer, 9 purines) with identical purine content but eluting 10 minutes apart. Using MSE data, we were able to rule out AAAAACAUGUUGCCGp as a false positive and confirm the identification of AUACAUUUGACAAAGp. This demonstrates the importance of strategically using both accurate-mass matching and fragmentation spectra to aid the unambiguous identification of components resulting from digested mRNA sequences. Additionally, our observations have led us to recognize that the retention of digested mRNA components may not be as predictable as first thought and that there is an urgent need for the interactions between oligonucleotides and chromatographic stationary phases to be further studied and modeled.
Lastly, we manually estimated sequence coverage by comparing the matched, manually validated digest components to the mRNA sequence. A preliminary estimate of sequence coverage for the 401 initial matches produced a coverage value of approximately 76%. When the rigorously validated matches were checked against the mRNA sequence, a coverage value of ~22% was obtained. Many of the observed digestion components mapped to more than one location in the luciferase mRNA sequence (Table 2) and the redundancy was expected because there are only four unique residues in a modified or fully modified nucleic acid sequence.
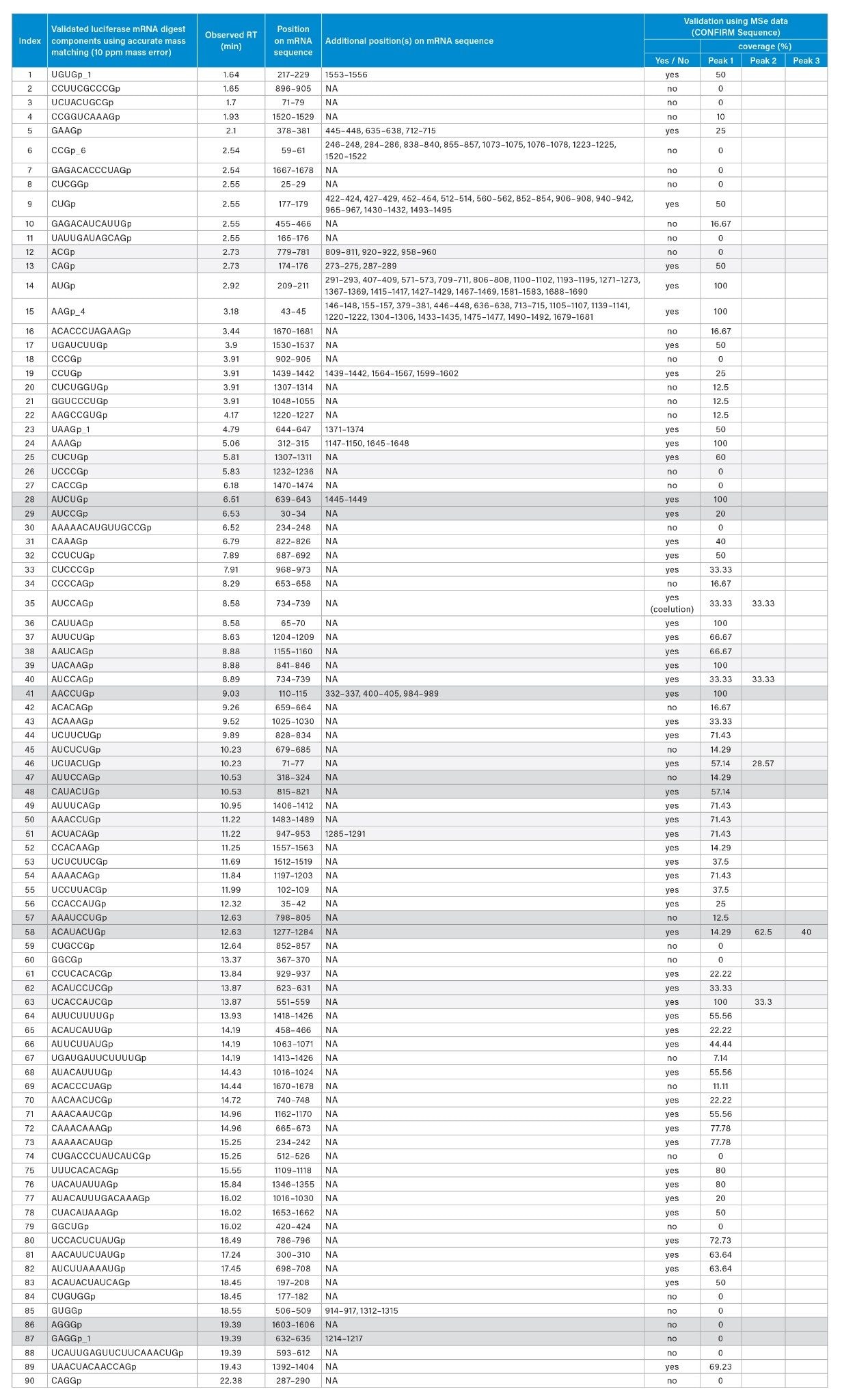 Table 2: Identification and validation of luciferase mRNA digest components based on accurate-mass matching and further validation using the waters_connect CONFIRM Sequence application and collected MSE spectra.
Table 2: Identification and validation of luciferase mRNA digest components based on accurate-mass matching and further validation using the waters_connect CONFIRM Sequence application and collected MSE spectra.
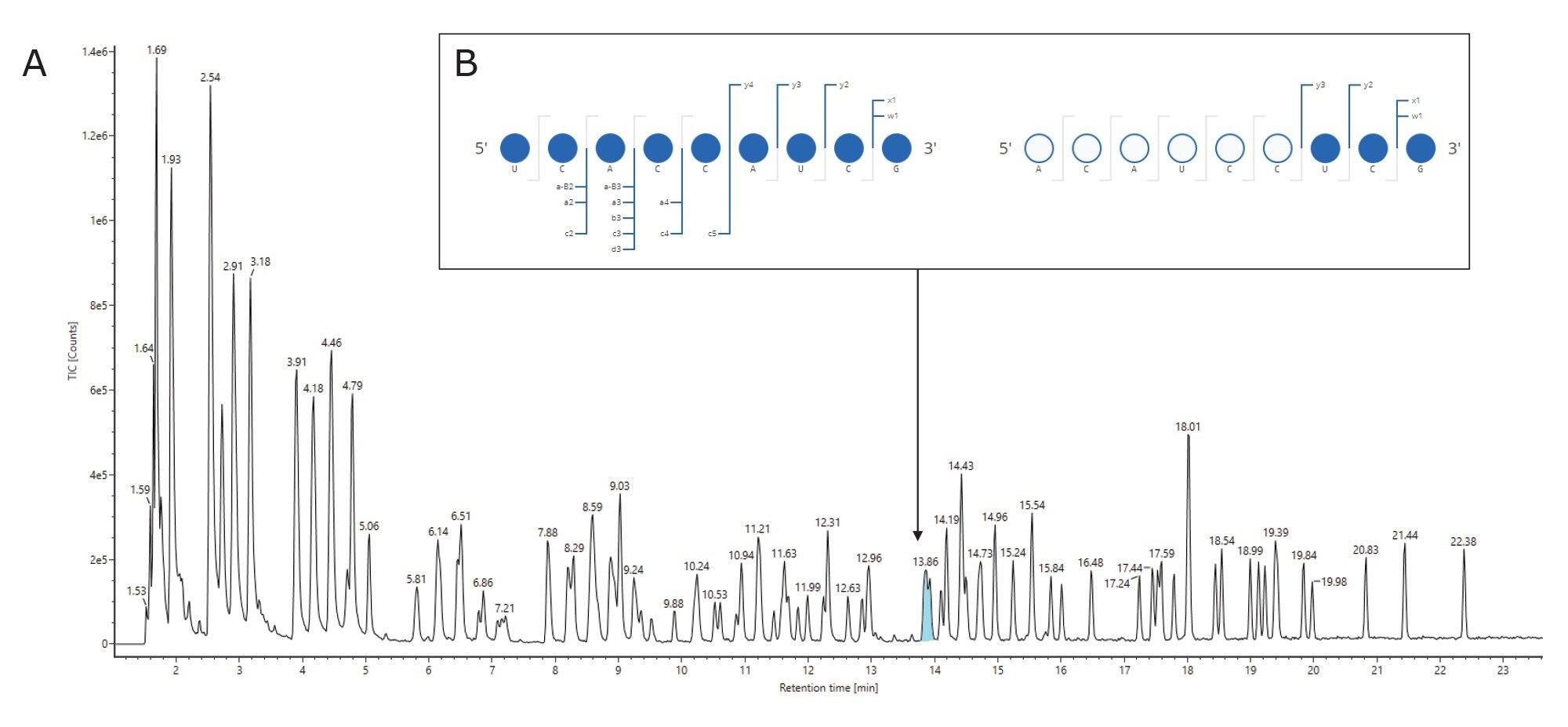 Figure 4: (A) Digested fragment components at position 623–631 (ACAUCCUCGp) and 551–559 (UCACCAUCGp) are predicted from RNAse T1 digest and are assigned to the same RT peak in the TIC. It is not possible to determine the correct assignment using intact mass information. (B) MSE data from the same injection can be used to elucidate the correct sequence for this assignment. Using the waters_connect CONFIRM Sequence application, high energy fragment ions are predicted using McLucky annotation11 for each sequence and matched to isotope clusters of the integrated raw data via a bespoke algorithm. The software presents confirmed fragment ions on a Dot-Map to quickly assess the sequence coverage.
Figure 4: (A) Digested fragment components at position 623–631 (ACAUCCUCGp) and 551–559 (UCACCAUCGp) are predicted from RNAse T1 digest and are assigned to the same RT peak in the TIC. It is not possible to determine the correct assignment using intact mass information. (B) MSE data from the same injection can be used to elucidate the correct sequence for this assignment. Using the waters_connect CONFIRM Sequence application, high energy fragment ions are predicted using McLucky annotation11 for each sequence and matched to isotope clusters of the integrated raw data via a bespoke algorithm. The software presents confirmed fragment ions on a Dot-Map to quickly assess the sequence coverage.
Conclusion
In the present work, we established a robust analytical workflow for oligo-mapping of synthetic mRNA using IP-RPLC and MS.
- Synthetic mRNAs were reproducibly digested using RNase T1, starting with as little as 10 µg of material, and injected without additional sample clean-up onto an ACQUITY Premier Oligonucleotide BEH C18 (2.1 x 150 mm, 300 Å, 1.7 µm) Column
- High chromatographic resolution was achieved using ion-pairing reversed-phase chromatography on an ACQUITY Premier LC such that digest components could be readily separated from incompletely digested mRNA and residual enzyme and efficiently detected with a BioAccord ACQUITY RDa Detector
- Annotated mRNA digest chromatograms were generated based on accurate-mass matching as facilitated by in silico mRNA digestion calculations and the application of waters_connect/UNIFI Scientific Libraries
- Assigned sequences for digested components were further validated based on MSE spectra using the waters_connect CONFIRM Sequence application. In addition, Dot-Map visualization was used to quickly check the fragment ion coverage of potential assignments
With this work, it was our intent to establish the chromatographic, detection, and data interpretation approaches that would be needed to facilitate the bottom-up characterization of an mRNA molecule. RNase T1 digestion was applied only as a first example and a proof of concept on establishing a workflow for data collection and analysis. That said, there is ample opportunity to more comprehensively probe a given mRNA structure by (1) using multiple, different nucleases to generate orthogonal and additive sequence mapping information and (2) adopting a multiplexing approach for data acquisition. These aspects, aimed at achieving comprehensive sequence coverage, will be explored in future work.
References
- Xu, S.; Yang, K.; Li, R.; Zhang, L., mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. International Journal of Molecular Sciences 2020, 21 (18), 6582.
- Brenner, S.; Jacob, F.; Meselson, M., An Unstable Intermediate Carrying Information From Genes to Ribosomes for Protein Synthesis. Nature 1961, 190 (4776), 576–581.
- Weide, B.; Pascolo, S.; Scheel, B. Derhovanessian, E.; Pflugfelder, A.; Eigentler, T. K. Pawelec, G.; Hoerr, I.; Rammensee, H. G.; Garbe, C., Direct Injection of Protamine-Protected Mrna: Results of a Phase 1/2 Vaccination Trial in Metastatic Melanoma Patients. J Immunother 2009, 32 (5), 498–507.
- Jiang, T.; Yu, N.; Kim, J.; Murgo, J.-R.; Kissai, M.; Ravichandran, K.; Miracco, E. J.; Presnyak, V.; Hua, S., Oligonucleotide Sequence Mapping of Large Therapeutic mRNAs via Parallel Ribonuclease Digestions and LC-MS/MS. Anal. Chem. 2019, 91 (13), 8500–8506.
- Plumb, R. S.; Johnson, K. A.; Rainville, P.; Smith, B. W.; Wilson, I. D.; Castro-Perez, J. M.; Nicholson, J. K., UPLC/MSE; A New Approach for Generating Molecular Fragment Information for Biomarker Structure Elucidation. Rapid Communications in Mass Spectrometry 2006, 20 (13), 1989–1994.
- Packer, M.; Gyawali, D.; Yerabolu, R.; Schariter, J.; White, P., A Novel Mechanism for the Loss of mRNA Activity in Lipid Nanoparticle Delivery Systems. Nat. Commun. 2021, 12 (1), 6777.
- Goyon, A.; Scott, B.; Kurita, K.; Maschinot, C.; Meyer, K.; Yehl, P.; Zhang, K., On-line Sequencing of CRISPR Guide RNAs and Their Impurities via the Use of Immobilized Ribonuclease Cartridges Attached to a 2D/3D-LC–MS System. Anal. Chem. 2021.
- Guo, L.; Worth, A. J.; Mesaros, C.; Snyder, N. W.; Glickson, J. D.; Blair, I. A., Diisopropylethylamine/Hexafluoroisopropanol-Mediated Ion-Pairing Ultra-High-Performance Liquid Chromatography/Mass Spectrometry for Phosphate and Carboxylate Metabolite Analysis: Utility for Studying Cellular Metabolism. Rapid Commun Mass Spectrom 2016, 30 (16), 1835–1845.
- Birdsall, R. E.; Gilar, M.; Shion, H.; Yu, Y. Q.; Chen, W., Reduction of Metal Adducts in Oligonucleotide Mass Spectra in Ion-Pair Reversed-Phase Chromatography/Mass Spectrometry Analysis. Rapid Commun Mass Spectrom 2016, 30 (14), 1667–1679.
- Fountain, K.; Gilar, M.; Budman, Y.; Gebler, J., Purification of Dye-Labeled Oligonucleotides by Ion-Pair Reversed-Phase High-Performance Liquid Chromatography. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences 2003, 783, 61–72.
- McLuckey, S. A.; Van Berkel, G. J.; Glish, G. L., Tandem Mass Spectrometry of Small, Multiply Charged Oligonucleotides. J. Am. Soc. Mass Spectrom. 1992, 3 (1), 60–70.
Acknowledgments
We would like to thank our colleagues at Waters for their valuable contribution to this work: Ana-Maria Rotaru, Emanuela Petreanu, Claudia Florea, Dave Jackson and Simon Jones. Many thanks to our collaborators at New England Biolabs: Bijoyita Roy, Siuhong Chan, Ivan R. Corrêa Jr., Erbay Yigit, and G. Brett Robb for providing luciferase mRNA and many thoughtful discussions.
720007669, June 2022