Direct Injection Screening Method for Per- and Polyfluoroalkyl Substances (PFAS) in Drinking Water Using the High Resolution Time of Flight Mass Spectrometer, the Xevo™ G3 QTof
Abstract
The widespread use of Per- and polyfluoroalkyl substances (PFAS) in industrial processing and manufacturing over the past few decades has led to global environmental and health concerns. As the number of these compounds continues to increase, it is becoming a challenging task to monitor PFAS using the traditional targeted methods. Screening workflow using High Resolution Mass Spectrometry (HRMS) is an ideal bridge for monitoring the regulated PFAS and detecting non-regulated PFAS. In this work, PFAS screening method is tested by direct injection of 10 µL of samples and standards. Samples were analyzed using an ACQUITY™ Premier UPLC™ System coupled to a Xevo G3 QTof Mass Spectrometer. Data was acquired in negative ionization mode using the MSE acquisition mode and processed using waters_connect™. The sensitivity and dynamic range of the Xevo G3 QTof were evaluated, with thirty out of the forty-six compounds detected at ≤5 ng/L, and a dynamic range for all compounds spanning over three orders of magnitude. Drinking water samples were analyzed, and PFHxA was detected in one sample with mass measurement accuracy of 0.8 ppm at 5 ng/L.
Benefits
- PFAS screening workflow using a direct injection approach on a LC coupled to a high-resolution time of flight mass spectrometer (QTof)
- High mass accuracy and high sensitivity at low ng/L concentrations of PFAS
- Analysis beyond targeted and regulated PFAS compounds where no standards are available for de novo identification
Introduction
Per- and polyfluorinated alkyl substances (PFAS) are a distinct class of man-made organic compounds in which the hydrogen atoms were partially or completely replaced by fluorine atoms. This C-F bond is the key to their remarkable properties, such as their water and lipid repellent, surfactant like properties and their high chemical and thermal stability against degradation. These properties, however, make PFAS highly persistent in the environment – and are known as forever chemicals.1-3 Since they were first reported in biological samples in the late 1960’s, the link between PFAS and different pathologies has been well established.6 Consequently, they are now considered a major environmental and health concern and therefore, the list of the PFAS of concern is constantly expanding.4-5,7-9
Targeted approaches for quantifying PFAS in different matrices are widely used.10-11 However, these methods are limited by low mass resolution and through their ability to only detect known compounds, for which a chromatographic retention time, precursor and product mass are required by analysing standards. The availability of these standards restricts targeted approaches to a small number of PFAS, around 40 per analysis method.11 Knowing the large number of listed PFAS compounds currently exceeds 15,000, using a screening method using high resolution mass spectrometry (HRMS) is an appealing approach to circumvent this challenge and increase the visibility in the studied samples to a wider range of PFAS related compounds.12 Data generated by HRMS can be qualitative and quantitative, with the information generated such as: the accurate mass and isotopic pattern allows for the generation of molecular formulas, a first step in de novo identification. In addition, the fragmentation profile in high collision energy scans allows further structural elucidation and putative identification of the component.13 The challenging requirements and detection limits, require the HRMS instrumentation and methodology to meet ng/L sensitivity levels.9,14
Sample preparation techniques such as solid phase extraction (SPE) are often recommended prior to analysis as means of sample cleanup and concentration. SPE techniques can, however, be biased towards the compounds with high adsorption affinity to the SPE cartridges. A number of PFAS related compounds are under proprietary protection, and potentially not listed in any of the watchlist databases.12,15 A complete identification of these compounds without using standards, would require complementary techniques such as Nuclear Magnetic resonance (NMR).16 The challenge with NMR, however, is that it requires a large sample volume and high concentrations. LC-HRMS direct injection methods are ideal for a non-biased inspection of the sample content providing putative identifications, based on accurate mass measurements, isotopic pattern, fragment ion information and mass defect filtering.13,17 The challenges within the industry highlight the need for a highly sensitive HRMS instrument that allow both screening, and the de novo annotation/identification.
In this study, an ACQUITY Premier UPLC System modified with an updated Isolator Column and PFAS kit coupled to a Xevo G3 QTof with the Waters™ PFAS library were used to demonstrate performance for screening PFAS in water samples.18
Experimental
Sample Preparation
All PFAS standards were purchased from Wellington laboratories. A stock solution in methanol at 10 ng/mL was prepared containing all the compounds listed in Table 1. From this stock solution serial dilutions were prepared at concentrations ranging from 0.5 ng/L to 5000 ng/L in methanol:water (1:1) + 0.1% Formic acid. Each concentration level was analysed in triplicate.
Water samples consisting of tap water, filtered water and MilliQ water were collected directly in the sample vials with no sample preparation, 10 µL of each sample were injected and analysed in triplicate.
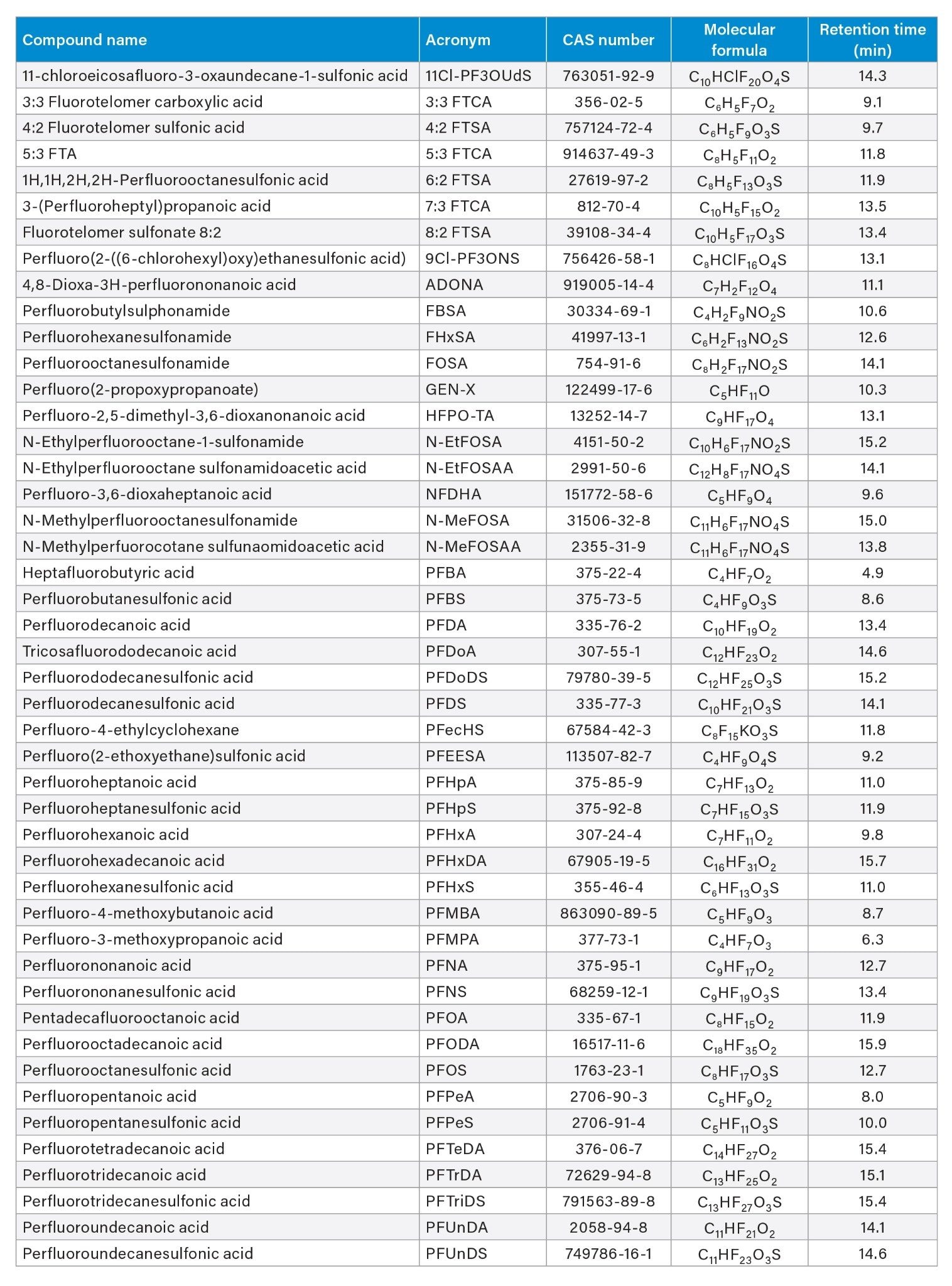 Table 1. List of PFAS standards present in the mixture and their retention times in minutes.
Table 1. List of PFAS standards present in the mixture and their retention times in minutes.
LC-MS Conditions
|
LC system: |
ACQUITY Premier Liquid Chromatography System modified with PFAS Kit (p/n: 205000588 and 205000589) |
|
Vials: |
Polypropylene autosampler vial (p/n: 186005219) with pre-slit cap (p/n: 186000305) |
|
Analytical column: |
ACQUITY Premier BEH™ C18, 1.7 µm, 2.1 x 100 mm, 90 Å Column (p/n:186009453) |
|
Isolator column: |
Atlantis™ Premier BEH C18 AX Isolater Column, 2.1 x 50 mm, 5 µm (p/n: 186009452) |
|
Column temperature: |
35 °C |
|
Sample temparature: |
6 °C |
|
Injection volume: |
10 µL |
|
Flow rate: |
0.3 mL/min |
|
Mobile phase A: |
95:5 water:methanol with 2mM ammonium acetate |
|
Mobile phase B: |
100% methanol with 2mM ammonium acetate |
LC Gradient Table
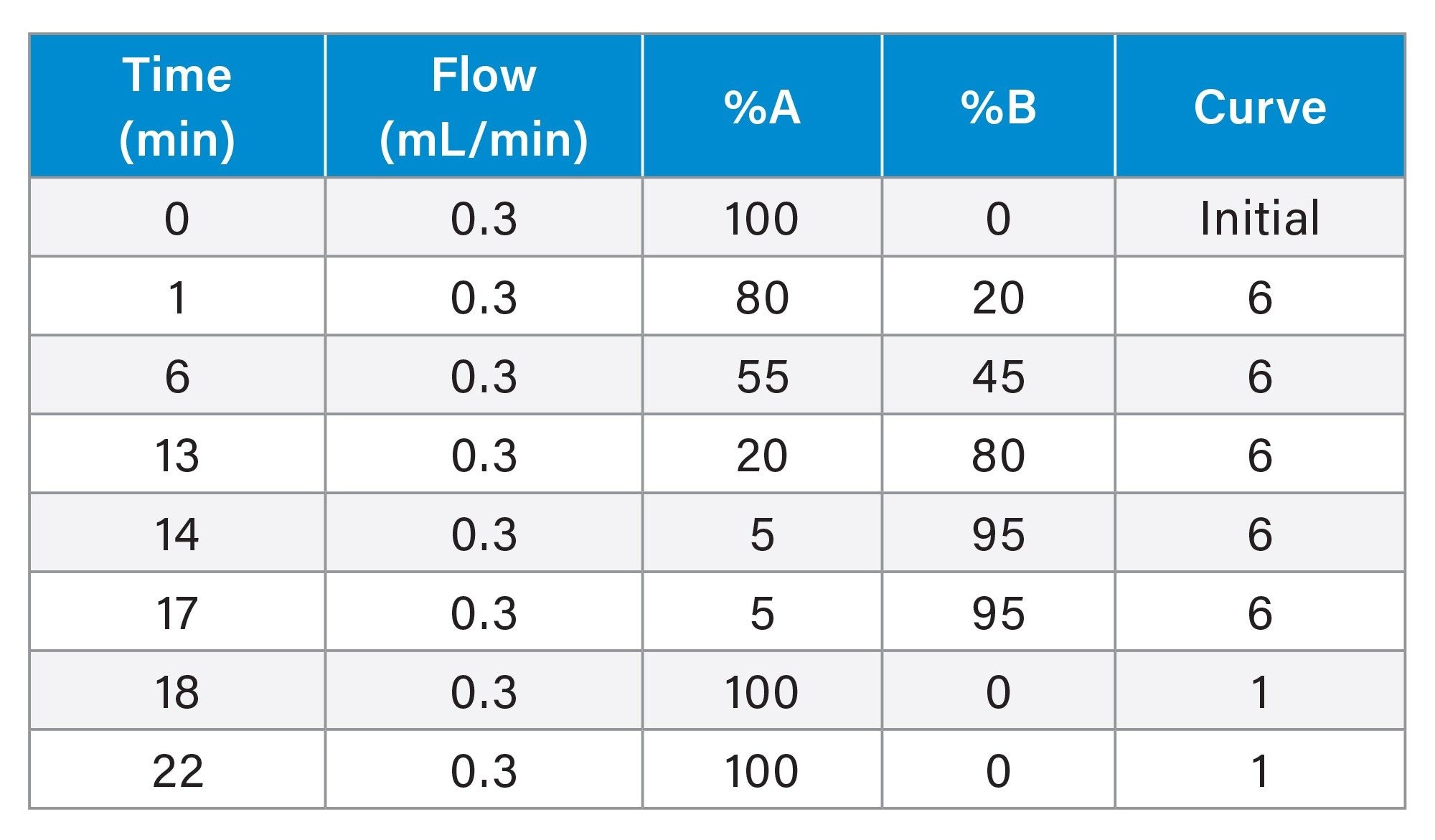
MS Conditions
|
MS system: |
Xevo G3 QTof |
|
|
Ionization mode: |
ESI- |
|
|
Mass range: |
m/z 50–1200 |
|
|
Acquisition rate: |
4 spectra per second (4 Hz) |
|
|
Lock mass: |
Leucine enkephalin (m/z 554.2620) |
|
|
Acquisition mode: |
MSE a data independent acquisition method |
|
|
Source conditions: |
||
|
Capillary voltage: |
0.5 kV |
|
|
Cone voltage: |
10 V |
|
|
Source temperature: |
100°C |
|
|
Desolvation temperature: |
250°C |
|
|
Cone gas: |
100 L/h |
|
|
Desolvation gas: |
600 L/h |
|
|
Source offset: |
30 V |
|
|
Collision energy |
||
|
Low collision energy: |
4 V |
|
|
High collision energy: |
ramp 20–70 V |
|
|
Transmission tune settings: |
||
|
StepWave RF: |
100 V |
|
|
Body gradient: |
5 V |
Software tools
Data acquisition was performed using waters_connect and data analysis performed within the UNIFI™ application.
Results and Discussion
Screening Workflow - Identification and Instrument Mass Accuracy
A solution containing forty-six PFAS standards at 2,000 ng/L was analysed by LC-HRMS using the MSE acquisition mode. The acquired data was then processed using the UNIFI Application screening workflow. This workflow included a mass defect filtering step to distinguish PFAS associated components. The generated list of components was screened against a Waters generated PFAS library.18 The components were identified based on accurate mass and fragmentation, with putative identifications accepted where the mass measurement accuracy was ≤3 ppm. Figure 1 depicts the extracted ion chromatogram (XIC) of forty-six identified PFAS in the standard mix at 2000 ng/L.
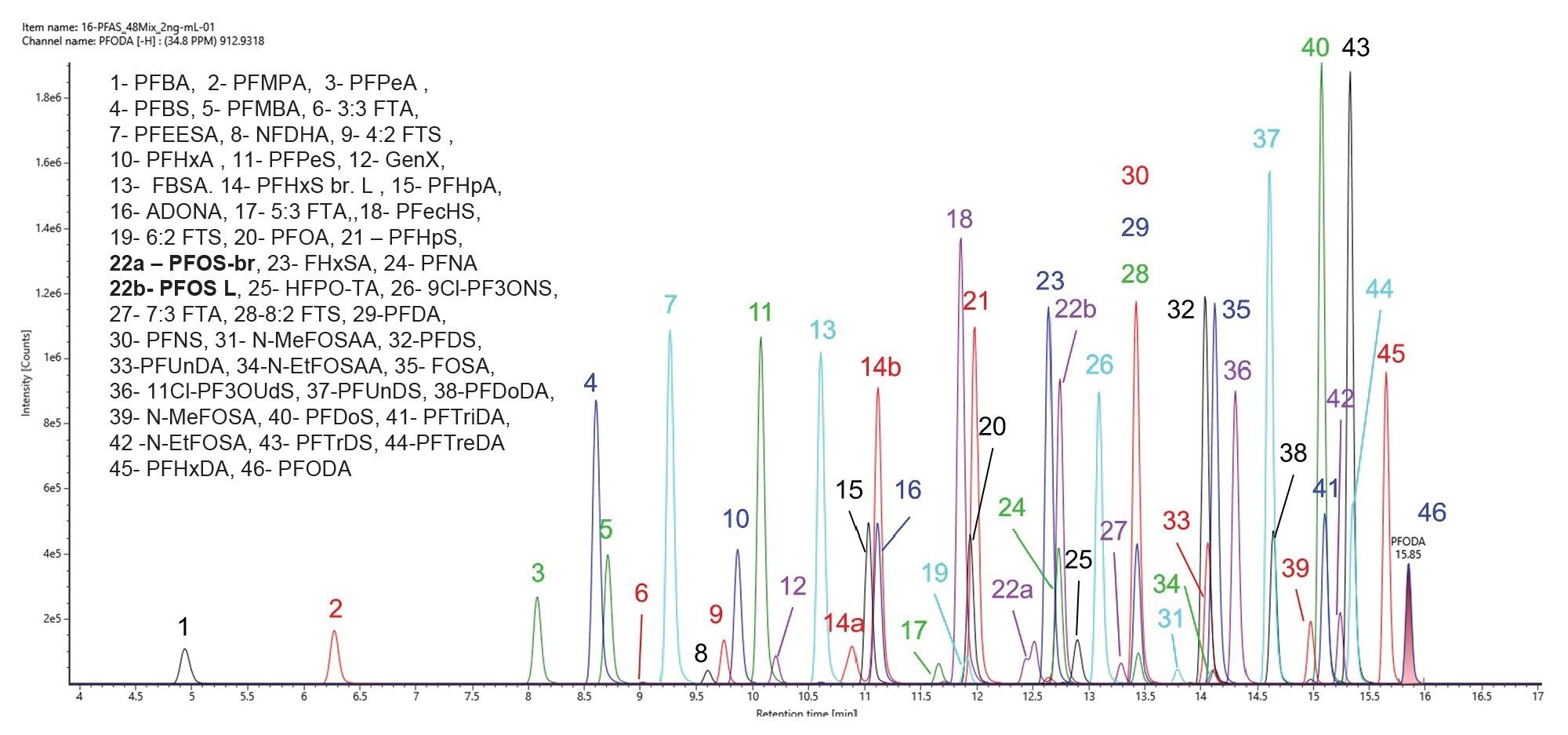 Figure 1. Extracted ion chromatogram (XIC) of forty-six PFAS standards identified in a solution at 2000 ng/L in 1:1 methanol:water containing 0.1% formic acid. The chromatographic method allowed the separation of linear and branched compounds. On this scale, only the visible isomers are indicated (such as branched and linear PFOS, compounds 22a and 22b, respectively).
Figure 1. Extracted ion chromatogram (XIC) of forty-six PFAS standards identified in a solution at 2000 ng/L in 1:1 methanol:water containing 0.1% formic acid. The chromatographic method allowed the separation of linear and branched compounds. On this scale, only the visible isomers are indicated (such as branched and linear PFOS, compounds 22a and 22b, respectively).
Figure 1 illustrates the elution order of the different classes of PFAS identified in the standard mix at 2000 ng/L with a mass measurement accuracy of ≤3 ppm. The PFAS LC kit (p/n: 205000588 and 205000589) is essential for distinguishing any residual lab contamination from the sample content as described by Adams et al 2023.10 The chromatographic method used allows for separation of the linear forms of PFAS analytes from their branched forms. For example, branched PFOS (PFOS-br) elutes earlier than the linear PFOS (PFOS-L) (Figure 1, peaks number 22a and 22b, respectively). Other linear/branched isomers such N-Methylperfuorocotane sulfunaomidoacetic acid (N-MeFOSAA) and N-Ethylperfluorooctane sulfonamidoacetic acid (N-EtFOSAA) are successfully separated and distinguished using this chromatographic method. Figure 2A depicts the extracted ion chromatogram (XIC) of N-MeFOSAA (linear and branched) and Figure 2B the mass spectra of linear N-MeFOSAA at low and high collision energy in a standard mix at 2000 ng/L (Figure 2B upper and lower panels, respectively).
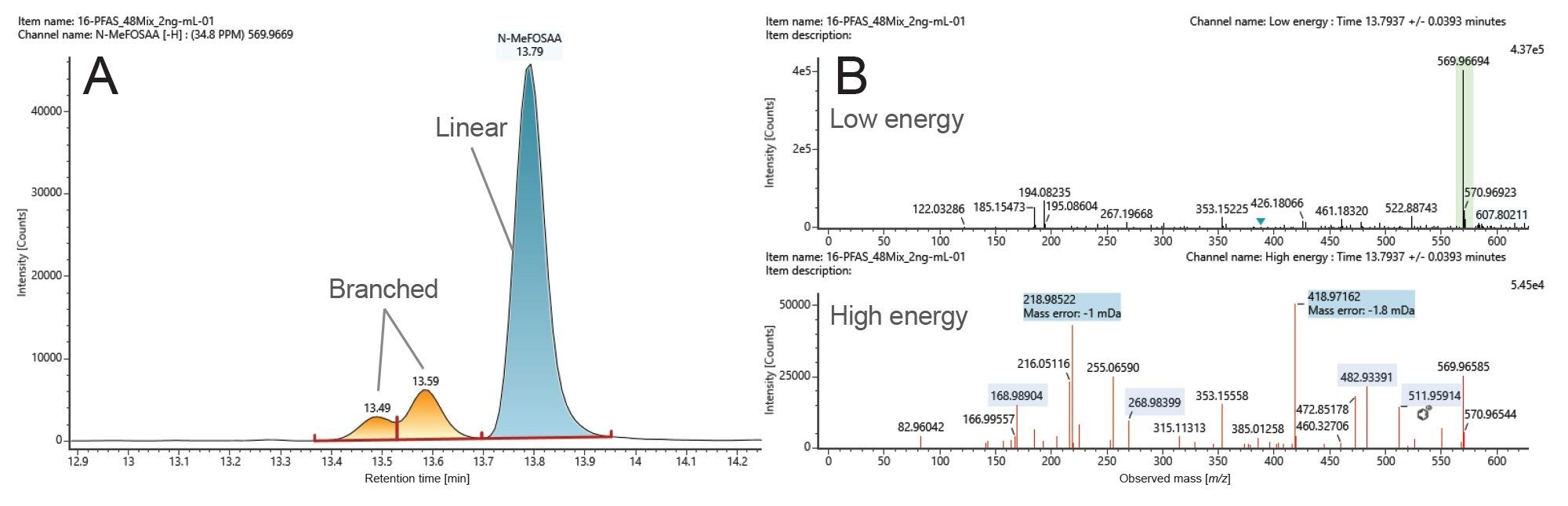 Figure 2. Identified N-Methylperfuorocotane sulfunaomidoacetic acid (N-MeFOSAA) in a standard mix at 2000 ng/L at -0.7 ppm. A: Extracted ion chromatogram (XIC) of N-MeFOSAA (linear and branched) and B: Mass spectra of linear N-MeFOSAA at low and high collision energy (upper and lower panels, respectively).
Figure 2. Identified N-Methylperfuorocotane sulfunaomidoacetic acid (N-MeFOSAA) in a standard mix at 2000 ng/L at -0.7 ppm. A: Extracted ion chromatogram (XIC) of N-MeFOSAA (linear and branched) and B: Mass spectra of linear N-MeFOSAA at low and high collision energy (upper and lower panels, respectively).
The XIC in figure 2A illustrates a main peak eluting at 13.8 minutes, and two minor peaks eluting at 13.5 minutes and 13.6 minutes (Figure 2B). The spectra shown in Figure 2B correspond to the low and high collision energy of the major peak eluting at 13.8 minutes, top and bottom panels, respectively. The low collision energy spectrum depicts a major ion at m/z 569.96694, identified as N-MeFOSSA with mass measurement accuracy of -0.7 ppm. The high energy spectrum depicts the different fragment ions generated from the base peak eluting at 13.8 minutes. Six out of these fragments correspond to the different bond breakage of linear N-MeFOSAA (Figure 3). Two out of the six fragments were not detected at 13.49 or 13.6 minutes, suggesting that the two ions correspond to the branched isomers N-MeFOSAA. These unique fragment ions at m/z 218.98525 and m/z 268.98256 correspond to [C4F9]- and [C5F11]-, respectively (Figure 3, highlighted in blue).
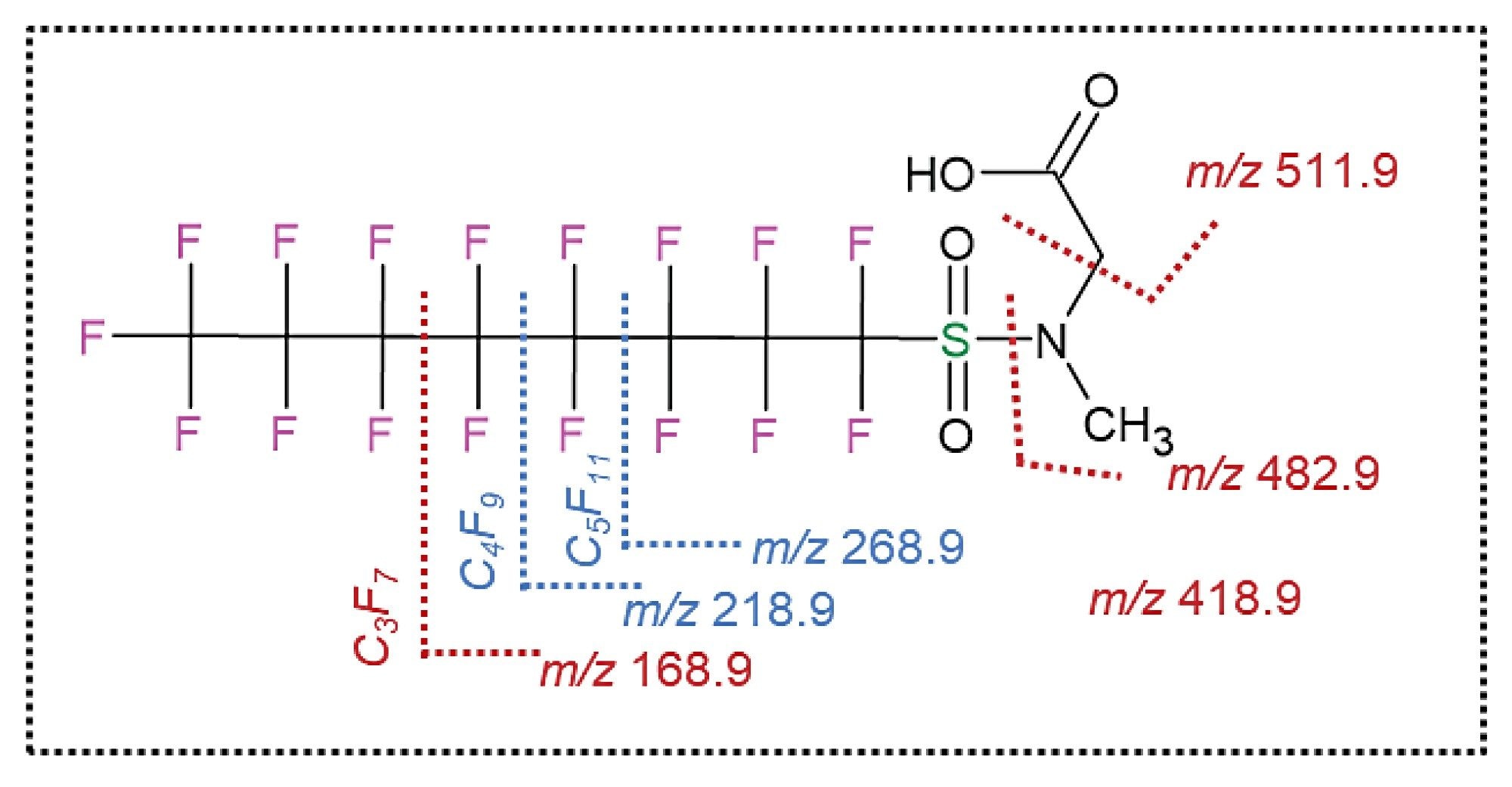 Figure 3. Structure of linear N-MeFOSAA with suggested fragmentation paths. Unique fragment ions for the linear isoform are indicated in blue, common fragment ions for the linear and branched are indicated in red.
Figure 3. Structure of linear N-MeFOSAA with suggested fragmentation paths. Unique fragment ions for the linear isoform are indicated in blue, common fragment ions for the linear and branched are indicated in red.
Linearity, sensitivity, and dynamic range
Using an LC equipped with non-PFAS leaching components such as the PFAS LC kit, is essential for reducing any residual lab contamination and ensuring any positively identified PFAS substances originated from the sample content.11 As PFAS are present in complex matrixes at low concentrations, the sensitivity and dynamic range of the Xevo G3 QTof was explored. Serial dilutions of the forty-six PFAS standard mix were prepared in concentrations ranging from 0.5 ng/L to 5,000 ng/L. The standards were analysed in triplicate and data processed using the UNIFI application within waters_connect. Both screening and quantitative approaches were utilized for data assessment. Calibration curves were obtained by plotting the response of each identified analyte from the low energy scan versus its corresponding concentration and fitted by linear regression with 1/X weighting.
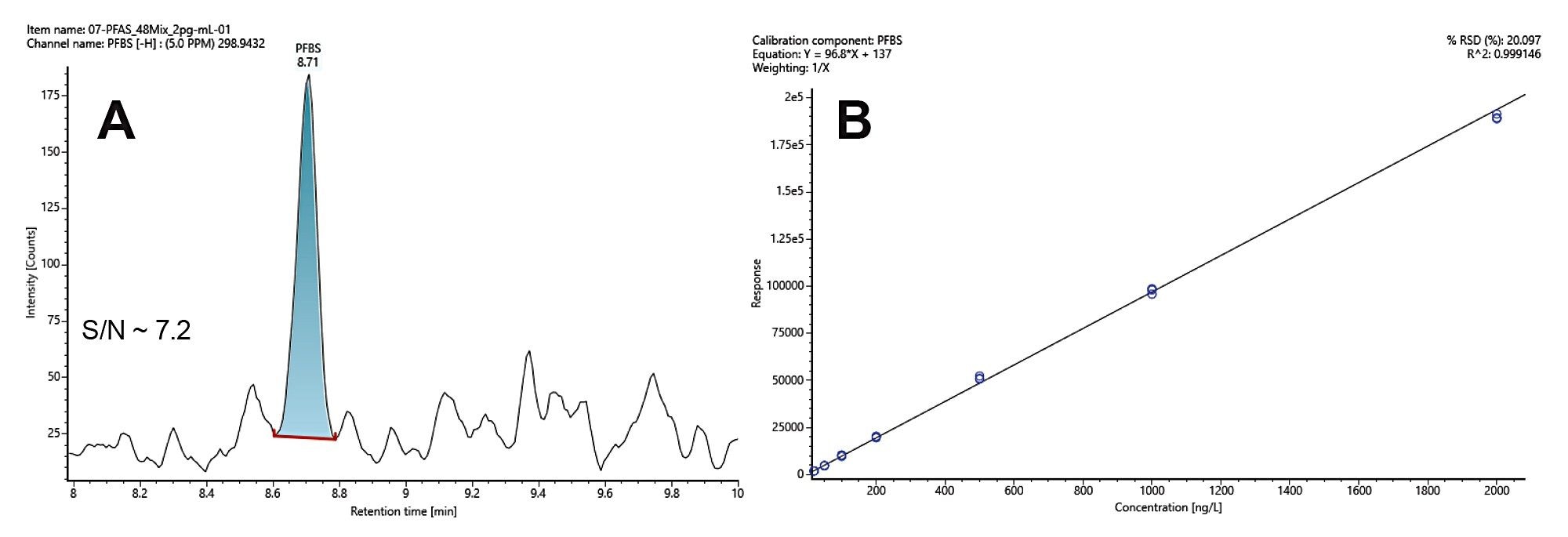 Figure 4. A: Extracted ion chromatogram of Perfluorobutanesulfonic acid (PFBS) at 2 ng/L. PFBS is identified at m/z 298.9432 with a mass measurement accuracy of ± 0.7 ppm and Signal-to-noise ratio at 7.2. The signal-to-noise is calculated using peak-to-peak method. B: calibration curve from the ion response at low collision energy. The linear regression is fitted with 1/X weighting with coefficient of determination R2=0.99915. PFBS response is linear from 2 ng/L to 2,000 ng/L.
Figure 4. A: Extracted ion chromatogram of Perfluorobutanesulfonic acid (PFBS) at 2 ng/L. PFBS is identified at m/z 298.9432 with a mass measurement accuracy of ± 0.7 ppm and Signal-to-noise ratio at 7.2. The signal-to-noise is calculated using peak-to-peak method. B: calibration curve from the ion response at low collision energy. The linear regression is fitted with 1/X weighting with coefficient of determination R2=0.99915. PFBS response is linear from 2 ng/L to 2,000 ng/L.
Figure 4A depicts an XIC for Perfluorobutanesulfonic acid (PFBS) at 2 ng/L with a signal-to-noise (S/N) ratio at 7.2, calculated using peak-to-peak method. PFBS was detected and identified at m/z 298.9432 with a mass measurement accuracy of ± 0.7 ppm. The response to PFBS is linear over the range of concentrations from 2 to 2,000 ng/L with a coefficient of determination (R2) 0.99915 (Figure 4B). It should be noted that the dynamic range of this approach covers three orders of magnitude allowing quantification of PFAS analytes with standards if they are available. For this analysis 10 µL of sample was injected on column, with the detection limit of PFBS at 2 ng/L or 0.02 pg on column (2 ppt), at a (S/N) ratio of >7.2. The linearity and sensitivity of all detected 46 compounds were investigated, with 30 of the compounds achieving a LLOD of ≤5 ng/L. The low limits of detections (LLOD), high limits of quantification (HLOQ), signal-to-noise (S/N), and the R2 for each of the studied compounds are summarised in table 2.
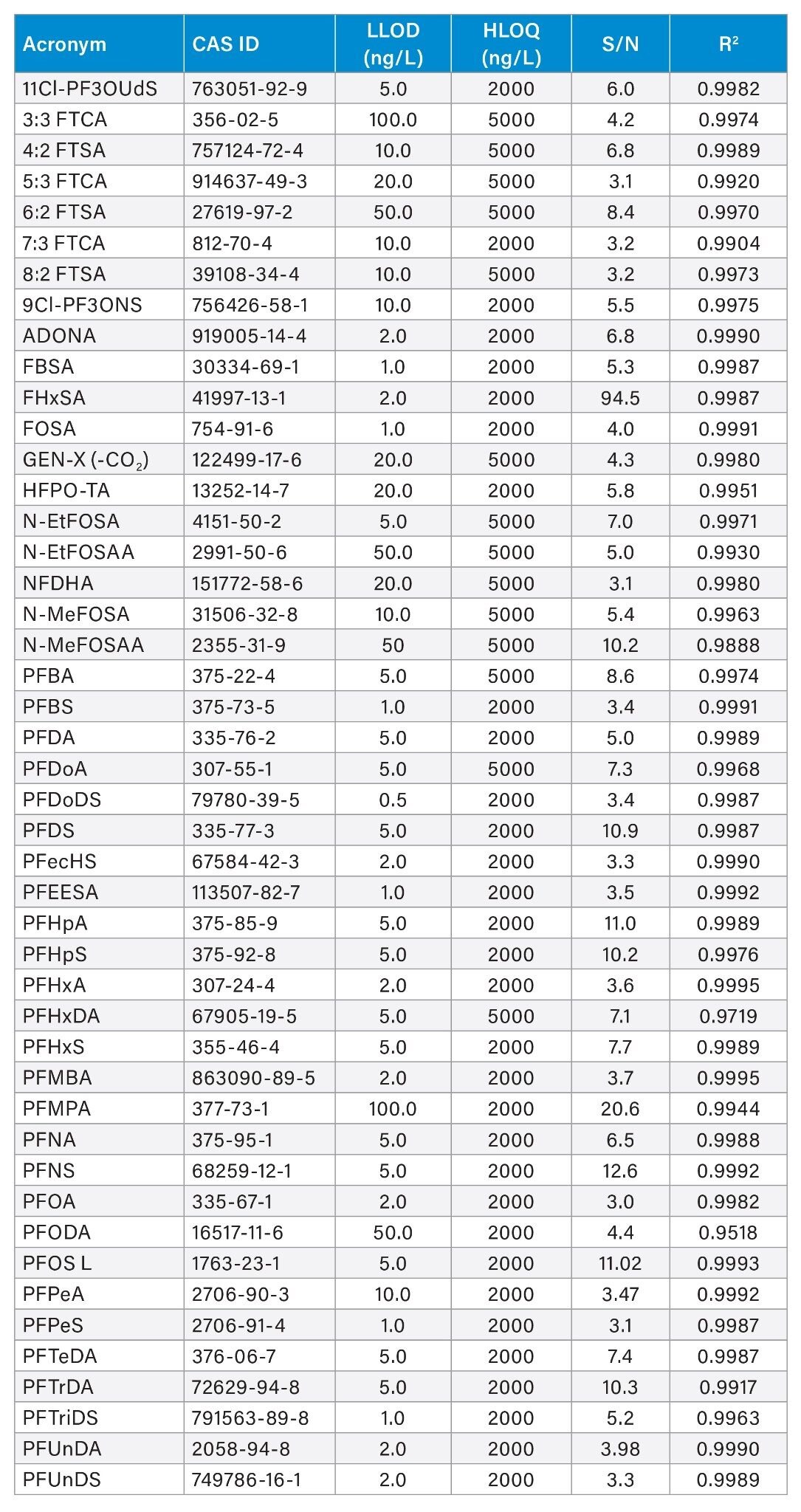 Table 2. Summary of the results of the forty-six standards detected, the low limit of detection (LLOD), the highest limit of quantification (HLOQ), the signal-to-noise ratio calculated using peak-to-peak method and the R2.
Table 2. Summary of the results of the forty-six standards detected, the low limit of detection (LLOD), the highest limit of quantification (HLOQ), the signal-to-noise ratio calculated using peak-to-peak method and the R2.
These results highlight the outstanding sensitivity of the Xevo G3 QTof with 30 compounds achieving a detection limit lower than or equal to 5 ng/L (0.05 pg on column). Among which PFBS, FBSA, FOSA and PFEESA with a LOD at 1 ng/L (0.01 pg on column), and PFHxA and ADONA at 2 ng/L (0.02 pg on column). Several of the values are lower than the method detection limits (MDL) requested by regulatory bodies where sample preparation and consequently sample concentration are required.14 Out of the forty-six compounds, four have a LLOD at 50 ng/L and two at 100 ng/L. Noteworthy, the analysis method used for this study is favouring labile compounds, and these limits of detection can be improved by modifying some certain source parameters if a targeted analysis is required.19 The linearity of the PFAS substances was demonstrated over three orders of magnitude with a minimum of six points on the calibration curve and an % RSD for the triplicate injections of <10%. All 46 compounds were shown to have a linear response and produced quantification curves with an R2 value of >0.99.
Detection and Quantitation of PFAS in Water Samples
The screening methodology was employed for the direct analysis of tap water, filtered drinking water, and MilliQ water, collected at source directly into the analysis vial with no sample preparation or clean-up. Out of the 46 PFAS compounds screened PFHxA was putatively identified within the tap water sample. PFHxA was identified with a mass measurement accuracy of -0.3 ppm and quantified at a concentration level of at 5 ng/L, where the lowest limit of detection of PFHxA had been determined at 2 ng/L (Figure 5).
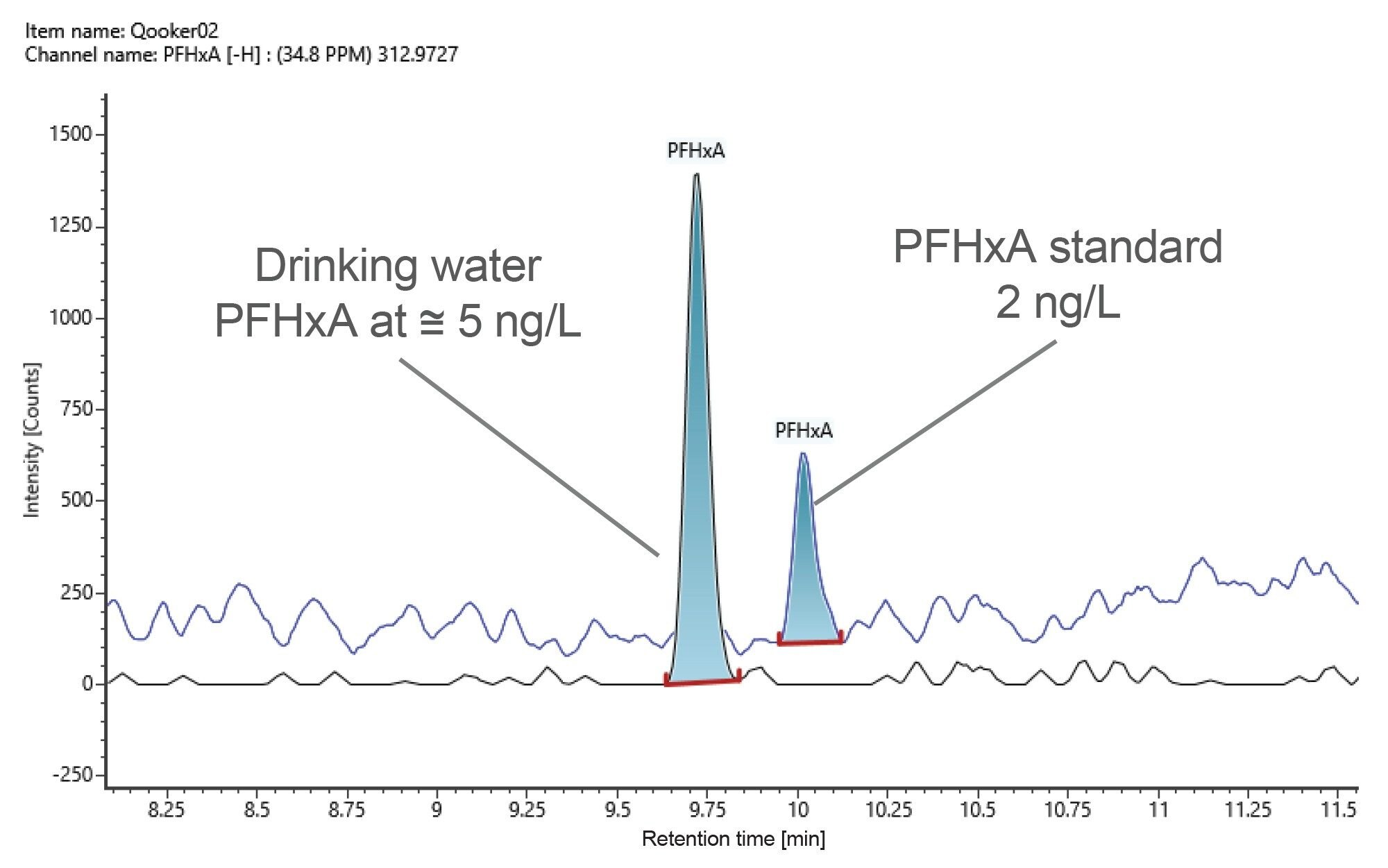 Figure 5. An overlay of extracted ion chromatograms of PFHxA in a standard solution and drinking water sample at 2 ng/L and at 5 ng/L, respectively. PFHxA was identified in drinking water with mass measurement accuracy of -0.3 ppm.
Figure 5. An overlay of extracted ion chromatograms of PFHxA in a standard solution and drinking water sample at 2 ng/L and at 5 ng/L, respectively. PFHxA was identified in drinking water with mass measurement accuracy of -0.3 ppm.
Conclusion
PFAS streamlined screening workflow using the UNIFI application in waters_connect coupled with the Xevo G3 QTof sensitivity were used for quantification of PFAS in water samples. The UNIFI screening and discovery workflows offer numerous options from mass defect filtering to online database and library searches. The data set generated was used for targeted quantitative approach and can be mined retrospectively for identification and discovery workflows using different library sources. The high sensitivity of the Xevo G3 QTof instrument helps circumvent the limitations of sample preparation using SPE and the need for commercially available standards for identification using conventional targeted methods. To increase sensitivity there is also the option of increasing injection volume.
References
- Giesy J P and Kannan K Global Distribution of Perfluorooctane Sulfonate in Wildlife. Environ. Sci. Technol. 35: 1339–1342, 2001.
- Pan C G, Liu Y S, Ying G G. Perfluoroalkyl Substances (PFASs) in Waste Water Treatment Plants and Drinking Water Treatment Plants: Removal Efficiency and Exposure Risk. Water Res. 106, 562–570, 2016.
- Podder A, Sadmani A H M A, Reinhart D, Chang N B, Goel R. Per and Polyfluoroalkyl Substances (PFAS) as a Contaminant of Emerging Concern in Surface Water: A Transboundary Review of their Occurrences and Toxicity Effects. J. Hazard. Mater. 419: 126361, 2021.
- Travis D R. Evidence that There are Two Forms of Fluoride in Human Serum. Nature 27: 1050–1051, 1968.
- Hansen K J, Clemen L A. Ellefson M E., Johnson H O. Compound-specific Quantitative Characterisation of Organic Fluorochemicals in Biological Matrices. Environ. Sci. Technol. 35, 766–770, 2001.
- Dickman R A. and Aga D S. A Review of Recent Studies on Toxicity, Sequestration, and Degradation of Per- and Polyfluoroalkyl Substances (PFAS) J. Hazard. Mater. 436: 129120, 2022 and references within.
- European Union Reference Laboratory for Halogenated POPs in Feed and Food. Guidance Document on Analytical Parameters for the Determination of Per- and Polyfluoroalkyl Substances (PFAS) in Food and Feed. Version 1.2. May 2022.
- Stockholm Convention on Persistent Organic Pollutants (POPs), 2019, Accessed, September 2021.
http://chm.pops.int/TheConvention/Overview/TextoftheConvention/tabid/2232/Default.aspx. - United States Environmental Protection Agency. Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Aqueous, Solid, Biosolids, and Tissue Samples by LC-MS/MS. 4th Draft method 1633, July 2023.
- Adams S, Dreolin N, Organtini K L, Hancock P. Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Animal Products with an Enhanced Sensitivity LC-MS/MS Method using Fish Reference Materials as a Case Study. Waters Application note. 720008108, 2023.
- Organtini K, Rosnack K, Hancock P. Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Accordance With EPA 1633 Part 1: Establishing and Assessing the Method. Waters Application note. 720008117, 2023.
- U.S. Environmental Protection Agency. CompTox Chemicals Dashboard. https://comptox.epa.gov/dashboard/chemical-lists/pfasmaster accessed November 2023.
- Charbonnet J A, McDonough C A, Xiao F, Schwichtenberg T, Cao D, Kaserzon S, Thomas K V, Dewapriya P, Place B J, Schymanski E L, Field J A, Helbling D E, Higgins C P. Communicating Confidence of Per- and Polyfluoroalkyl Substance Identification via High-Resolution Mass Spectrometry. Enviorn. Sci. Technol. Lett. 9: 473–481, 2022.
- United States Environmental Protection Agency. Method 533. Determination of Per and Polyfluoroalkyl Substances in Drinking Water by Isotope Dilution Anion Exchange Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry. November 2019.
- Suspect List of Possible Per- and Polyfluoroalkyl Substances (PFAS). NIST Public Data Repository. https://data.nist.gov/od/id/mds2-2387 accessed November 2023.
- Camdzic D, Dickman R A, Joyce A S, Wallace J S, Ferguson P L, Aga D S. Quantitation of Total PFAS Including Trifluoroacetic Acid with Fluorine Nuclear Magnetic Resonance Spectroscopy. Anal. Chem. 95: 5484–5488, 2023.
- Schymanski E L, Singer H P, Slobodnik J, Ipolyi I M, Oswald P, Krauss M, Schulze T, Haglund P, Letzel T, Grosse S, Thomaidis N S, Bletsou A, Zwiener C, Ibáñez M, Portolés T, de Boer R, Reid M J, Onghena M, Kunkel U, Schulz W, Guillon A, Noyon N, Leroy G, Bados P, Bogialli S, Stipaničev D, Rostkowski P, Hollender J. Non-target Screening with High-Resolution Mass Spectrometry: Critical Review Using a Collaborative Trial on Water Analysis. Anal. Bioanal. Chem. 407:6237–6255, 2015.
- Towhig M, Fujimoto G, Mohan A, Organtini K L, Rosnack S and Hird S. Approaches to Non-targeted Analyses of Per- and Polyfluoroalkyl Substances (PFAS) in Environmental Samples. Waters Application note. 720007184, 2021.
- Khoury-Hollins, H, Riba I, Kirk J. Optimization of Source and Transmission Parameters for a Mix of Labile and Stable Per – or Polyfluoroalkyl Substances (PFAS) Using the Xevo™ G3 QTof Mass Spectrometer. Waters Application note. 720008118, 2024.
720008198, January 2024