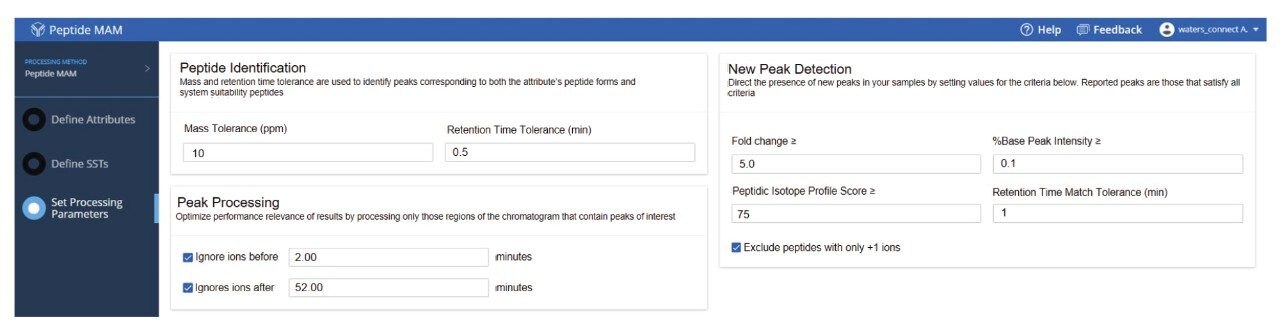
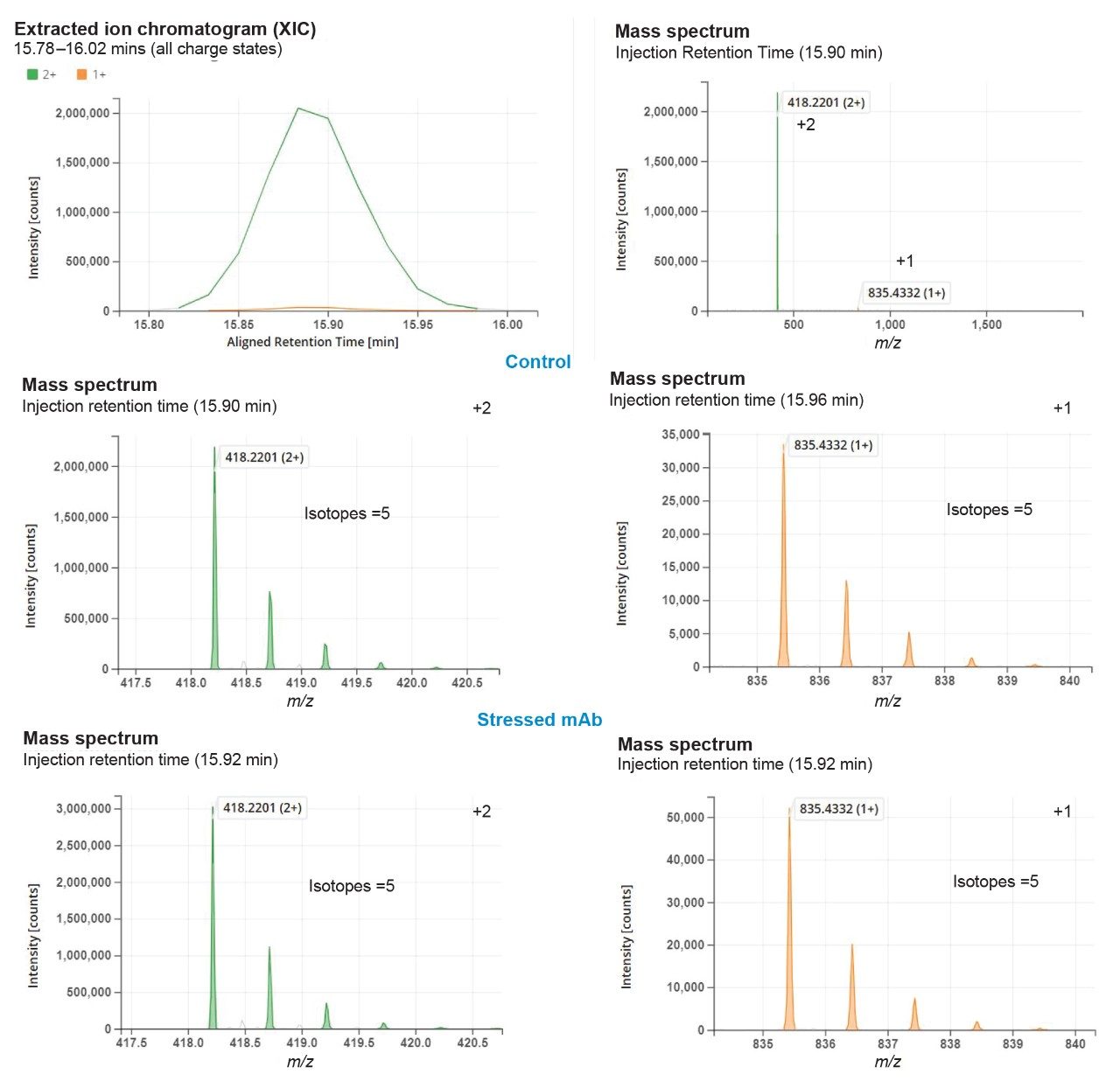
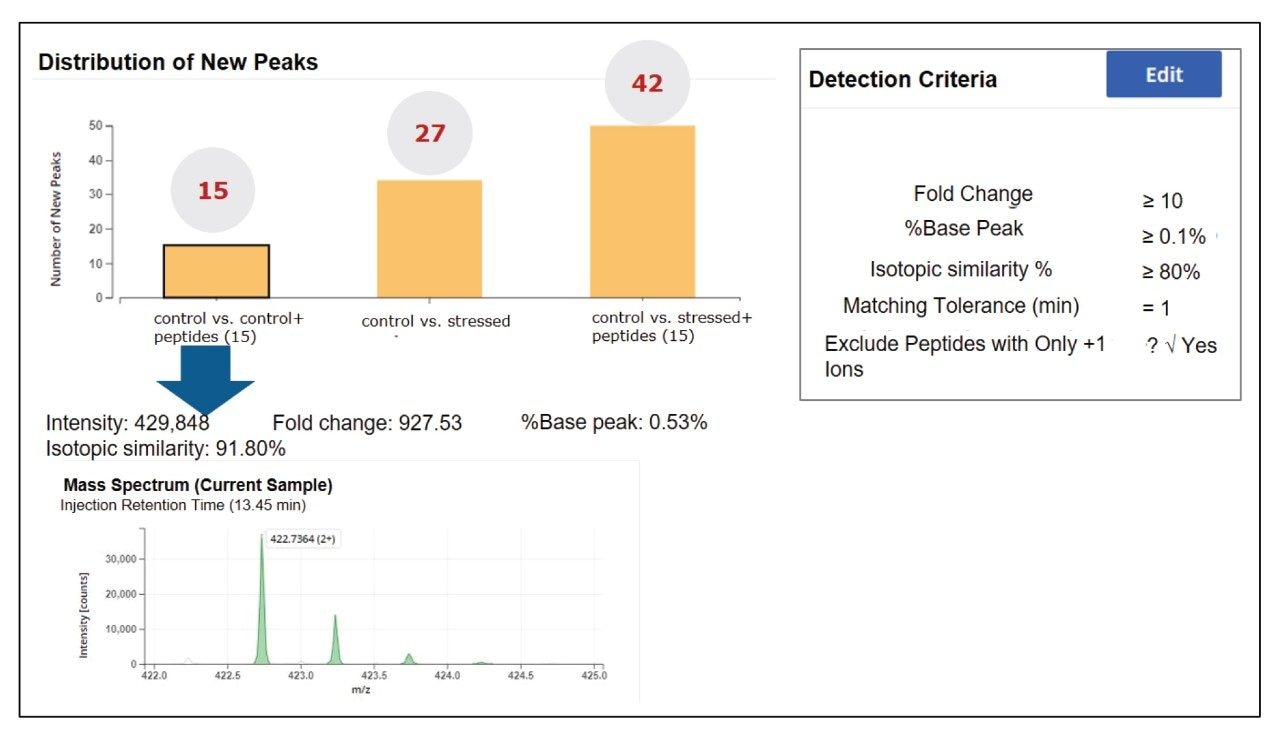
The NPD filtering criteria can include fold change, %base peak, %isotopic similarity, and retention time tolerance (min). During peak processing the fold change of a new peak is calculated relative to the MS intensity of the codetected peak in the reference/control sample. Any elevation or reduction in peak intensity beyond the thresholds will trigger this criterion for a new peak detection. Since fold-change is calculated relative to a reference sample, the process can standardize impurity level measurements across all analytical samples. In this study, as per industry practice, ≥10 was used as the default fold change.
The %base peak is another criterion used in NPD used to avoid false positive identifications from background ions and chemical noise. This is calculated relative to the most intense peptide’s MS response within an LC-MS chromatogram. In the example data for the forced degradation study (Figure 7) the unmodified VVSVLTVLHQDWLNGK peptide was determined as the base peak by the software. The study used a %base peak at a minimum of 0.1% level to set a lower limit for new peak detection.
To further minimize false positive identifications, an isotopic similarity score has been introduced into peptide MAM NPD data processing. The %isotopic similarity is calculated relative to the isotopic distribution of an ion with similar m/z. BioAccord data collected with intelligent data capture (IDC) has this score typically set to 75% or higher. The matching tolerance for retention time was maintained at 1 min and excluded all solvent ions and chemical noise by selecting “Exclude peptides with only +1 ions option”.
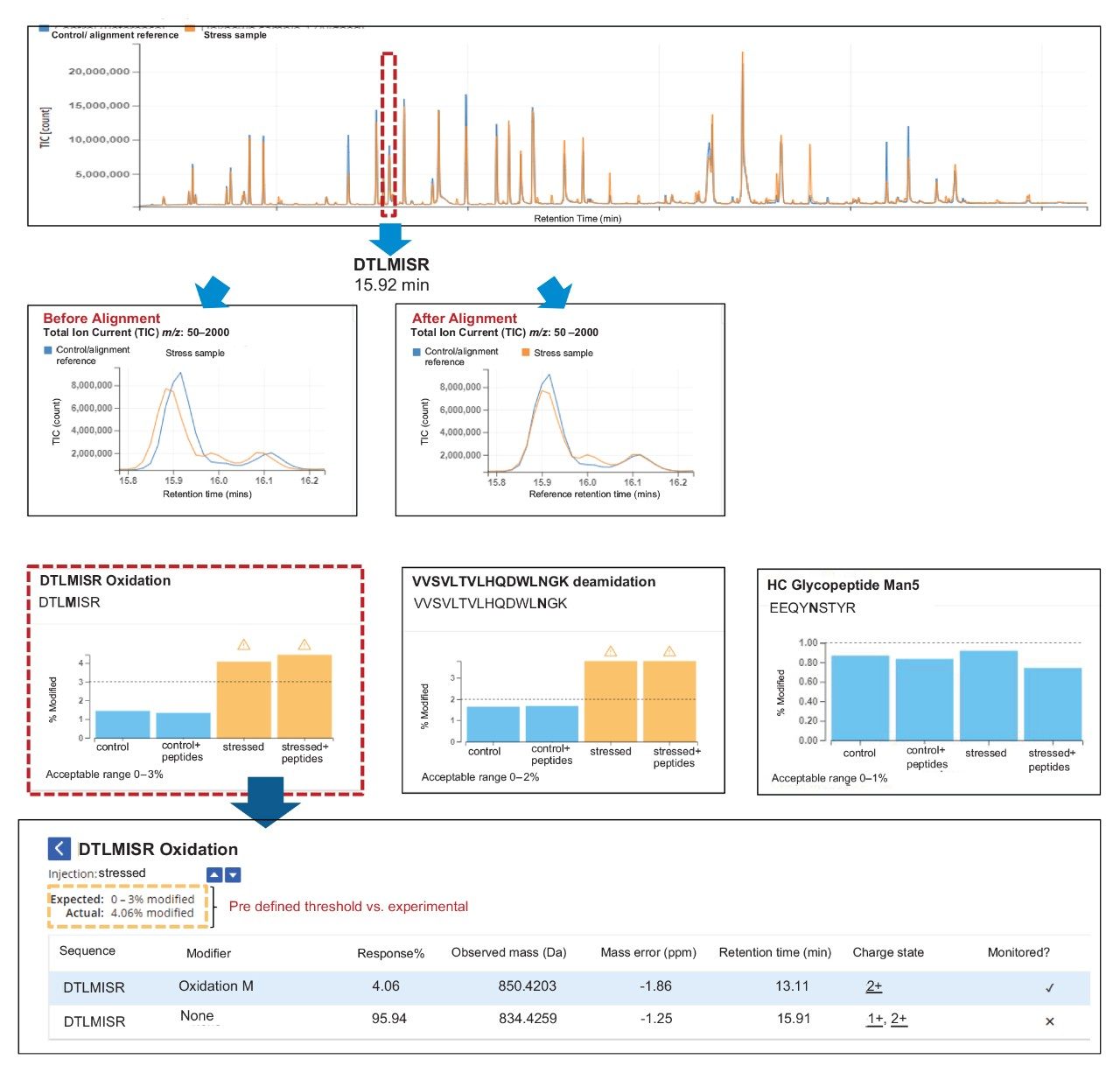
The NPD results (Figure 7) showed identification of 15 new peaks in spiked control sample that corresponded to the neutral mass of spiked heavy labeled peptides. In the stressed NISTmAb sample the number of new peak peaks was 27. These peaks contained modifications such as oxidations and deamidations, typical of previous studies of the NIST mAb. The stressed and spiked mAb sample should result in 42 (15 spike + 27 stressed) new peaks, matching our observations.
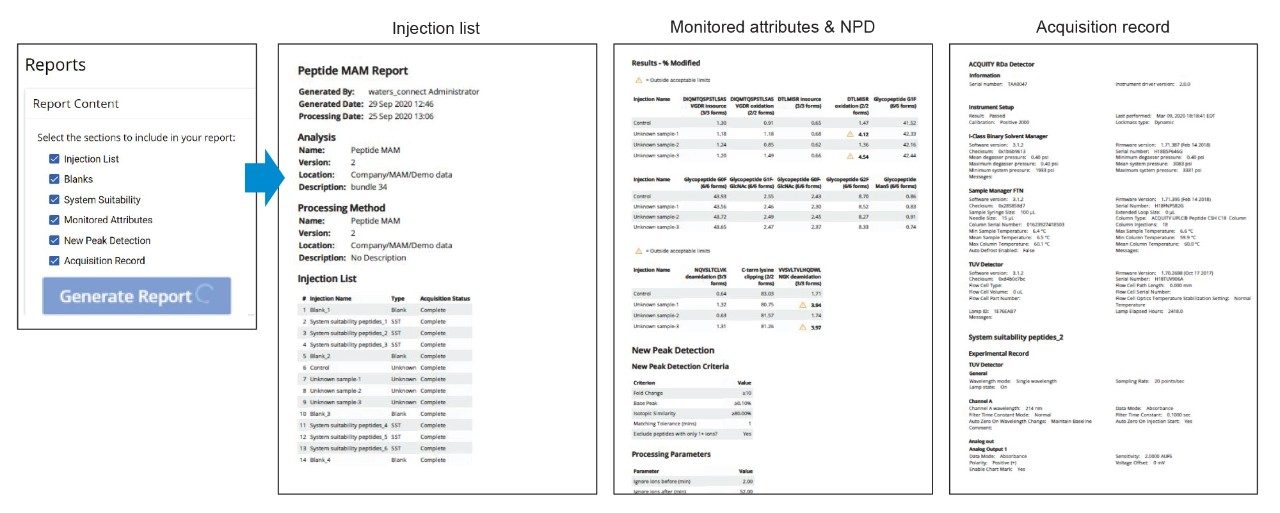
If needed, each new peak can be verified using a “review” option, enabling MS spectra to be displayed for the selected new peaks. Authorized users can accept or reject these new peaks prior to finalizing the results.