臨床研究のための血漿中のクロザピンおよびノルクロザピンの迅速分析
研究目的のみに使用してください。診断用には使用できません。
要約
大規模薬物動態試験や集団試験では、頑健でハイスループットの分析手順を利用できることが有利になります。ここでは、比較的少量のサンプルを使用し、シンプルで低コストの前処理手法を用いた、臨床研究の現場に適した分析法について説明します。次に、液体クロマトグラフィーとタンデム質量分析を用いたクロザピンおよびノルクロザピンの定量について説明します。
アプリケーションのメリット
- 実行時間が短く(1 サンプルあたり 72 秒)ハイスループットの機能
- クロマトグラフィーおよび質量検出器によって分析の選択性を実現
- 少量のサンプルを使用したシンプルで低コストの前処理
はじめに
大規模薬物動態試験や集団試験では、頑健でハイスループットの分析手順を利用できることが有利に働く可能性があります。
今回、アセトニトリル中の内部標準を使用したヒト血漿の除タンパク、続いて液体クロマトグラフィーとタンデム質量分析を使用してクロザピンおよびノルクロザピンを定量する、臨床研究に適した分析法について説明します。
ACQUITY UPLC™ I-Class フロースルーニードルシステムで Waters™ XBridge™ Premier BEH™ C18 カラムを使用すると、クロマトグラフィー溶出が 42 秒で完了し、注入間の時間が 72 秒になりました。Xevo™ TQ-S micro 質量分析計でのマルチプルリアクションモニタリング(MRM)によって分析種が検出されました(図 1)。
 図 1.ACQUITY UPLC I-Class システムと Xevo TQ-S micro 質量分析計
図 1.ACQUITY UPLC I-Class システムと Xevo TQ-S micro 質量分析計
実験方法
サンプル前処理
血漿キャリブレーターと品質管理物質は、BioIVT(英国、ウェストサセックス)から提供されたプールしたヒト血漿を使用して調製しました。濃縮ストック溶液は、Toronto Research Chemicals(カナダ、オンタリオ州)から提供されたクロザピン、ノルクロザピン、内部標準としての重水素標識クロザピンの認定粉末から調製しました。両方の分析種について、キャリブレーション範囲は 50 ~ 2,000 ng/mL でした。
サンプル抽出
3 倍量過剰の内部標準(2H4-クロザピンアセトニトリル溶液)を、マイクロ遠心分離チューブ内の 50 µL のサンプルに添加しました。マルチチューブボルテックスミキサーでチューブを 2,000 rpm で 1 分間攪拌してから、室温で 16,200 × g で 5 分間遠心分離しました。20 µL の上清を 2 mL の 96 ウェルプレートに移し、450 µL の 30%(v/v)メタノール(水溶液)を加えました。
UPLC 条件
|
システム: |
ACQUITY UPLC I-Class フロースルーニードル(FTN) |
|
ニードル: |
30 µL |
|
UPLC カラム: |
XBridge Premier BEH C18 UPLC、粒子径 2.5 µm、2.1 × 50 mm 内径(製品番号:186009827) |
|
インラインカラムフィルター: |
XBridge Premier BEH C18 UPLC、粒子径 2.5 µm、2.1 × 5 mm VanGuard™ FIT カートリッジ(製品番号:186009842) |
|
移動相(A): |
5 mM ギ酸アンモニウム含有 0.1%(v/v)ギ酸(水系) |
|
移動相(B): |
5 mM ギ酸アンモニウム含有 0.1%(v/v)ギ酸の 80/20(v/v)メタノール/イソプロパノール溶液 |
|
ニードル洗浄溶媒: |
0.1%(v/v)ギ酸含有メタノール/アセトニトリル/イソプロパノール/水 1/1/1/1(v/v)溶液 |
|
パージ溶媒: |
30%(v/v)メタノール(水系) |
|
シール洗浄溶媒: |
パージ溶媒と同じ |
|
カラム温度: |
45 ℃ |
|
注入量: |
2 µL |
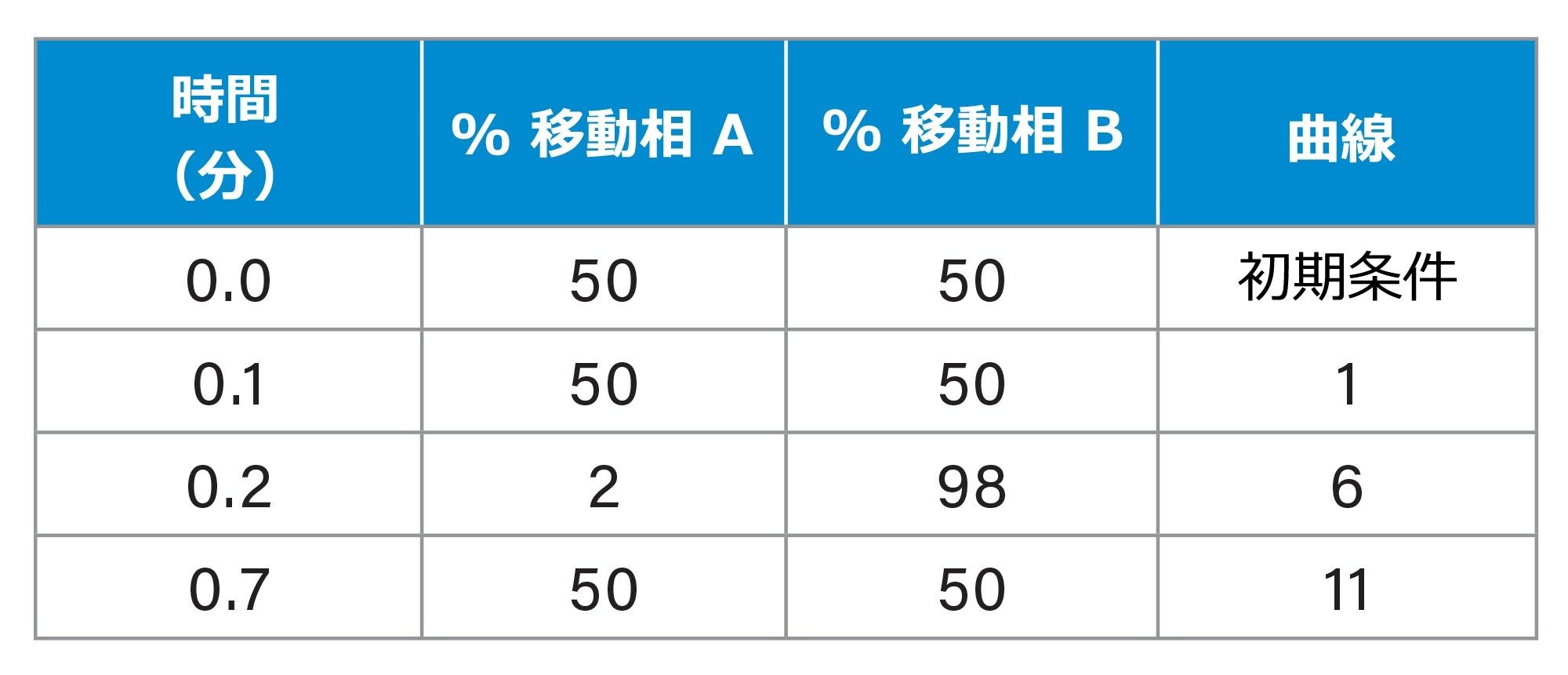 表 1.バイナリーソルベントマネージャーグラジエントテーブル
表 1.バイナリーソルベントマネージャーグラジエントテーブル
|
流速: |
0.6 mL/分 |
|
実行時間: |
0.7 分(注入間 1.2 分) |
MS 条件
|
システム: |
Xevo TQ-S micro |
|
四重極分解能: |
0.7 FWHM での MS1 および MS2(0.6 ~ 0.8 を許容) |
|
取り込みモード: |
マルチプルリアクションモニタリング(MRM)(表を参照) |
|
メソッドのイベント: |
MS 廃液へ転流(0.2 分未満および 0.5 分超) |
|
極性: |
エレクトロスプレーポジティブイオン化 |
|
キャピラリー電圧: |
3.00 kV |
|
イオン源温度: |
150 ℃ |
|
脱溶媒温度: |
600 ℃ |
|
脱溶媒ガス流量: |
1,000 L/時間 |
|
コーンガス流量: |
25 L/時間 |
|
チャンネル間、スキャン間および極性切替の遅延: |
自動 |
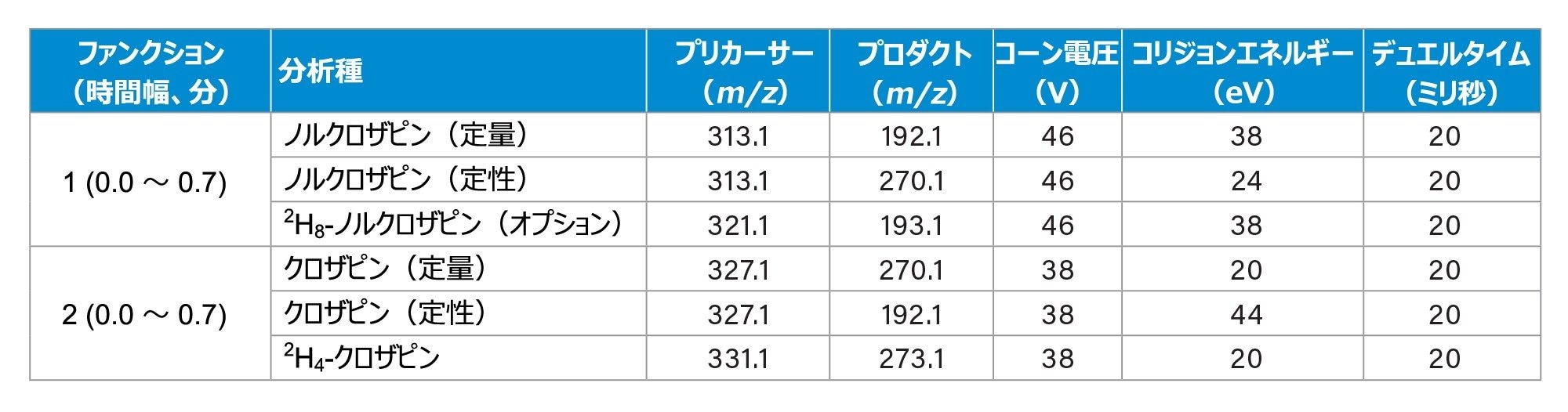 表 2.マルチプルリアクションモニタリングイオントランジション
表 2.マルチプルリアクションモニタリングイオントランジション
|
データ管理: |
MassLynx™ v4.2(TargetLynx™ アプリケーションマネージャー搭載) |
結果および考察
表 3.に開発した分析法の性能特性をまとめており、不精確さおよびバイアスに関する性能が優れていることが実証されています。
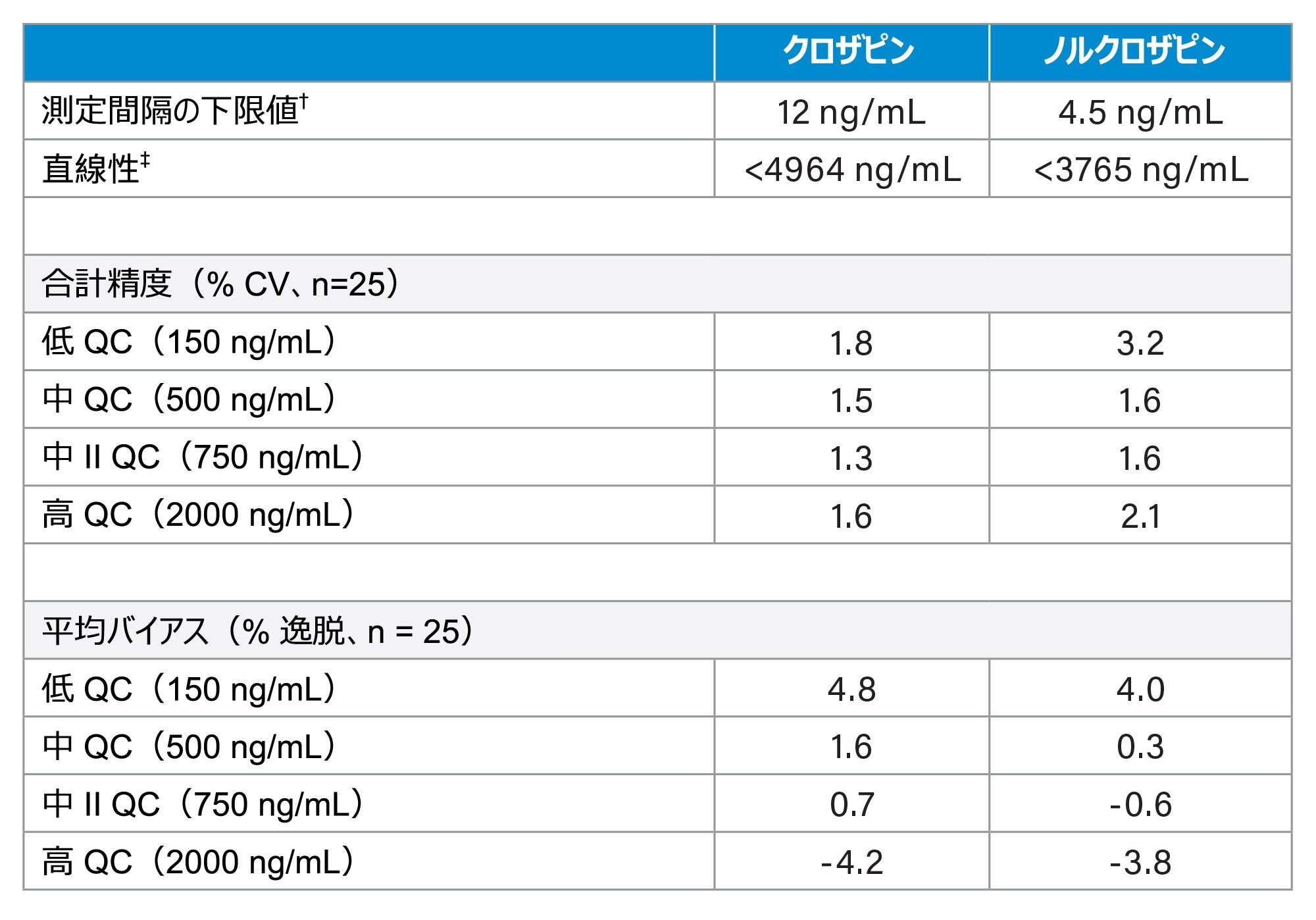 表 3.性能のサマリー
表 3.性能のサマリー†最低公称濃度を不精確さ 20% 以下(相対標準偏差(RSD))で測定可能で、公称濃度からの逸脱 15% 以下。
‡2 次多項式または 3 次多項式に有意な値がなく(p < 0.05)、r2 は 0.997 以上。
有機溶媒含有量が多い洗浄溶媒や移動相を使用すると、ブランクサンプルを高濃度サンプル(キャリブレーター最大濃度の約 2 倍)と連続して分析した場合の結果からわかるように(各条件ごとに n = 10、データは示していません)、キャリーオーバーがほとんどなくなりました(LLMI の 25% 未満)。
サンプルの希釈、低注入量、LC 溶出液の廃液への転流の組み合わせにより、クロザピンとノルクロザピンの安定したクロマトグラフィー分離および定量が可能になり、保持時間の偏差は予想保持時間から 0.36 秒以内で、実測濃度のばらつきは 1.2%(RSD)以下でした(図 2)。
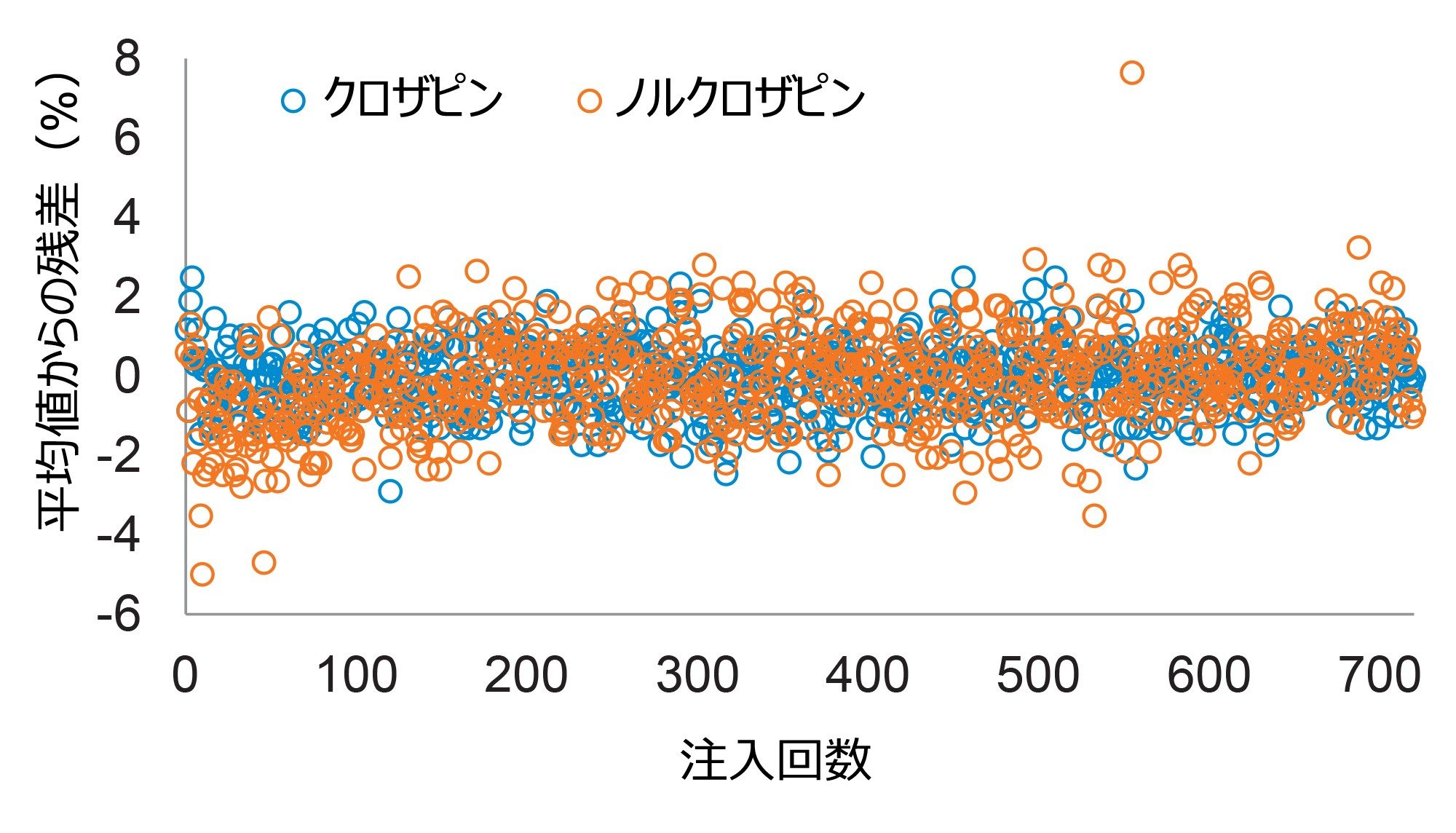 図 2.14 時間の連続動作によってクロザピンとノルクロザピンを定量したところ、長期間の分析にわたって検出器が安定していることがわかります。プールしたサンプル中の平均濃度および測定の不精確さ(%RSD)は、クロザピンでは 688 ng/mL(0.8%)、ノルクロザピンでは 680 ng/mL(1.2%)でした(n = 720)。
図 2.14 時間の連続動作によってクロザピンとノルクロザピンを定量したところ、長期間の分析にわたって検出器が安定していることがわかります。プールしたサンプル中の平均濃度および測定の不精確さ(%RSD)は、クロザピンでは 688 ng/mL(0.8%)、ノルクロザピンでは 680 ng/mL(1.2%)でした(n = 720)。
流す溶媒の有機溶媒含有量を 6 秒間で 48% 増加させたステップグラジエントでは、クロザピンとノルクロザピンが共溶出しました(図 3)。共溶出により、両化合物の定量に、単一の内部標準を使用することを評価する機会が得られました。
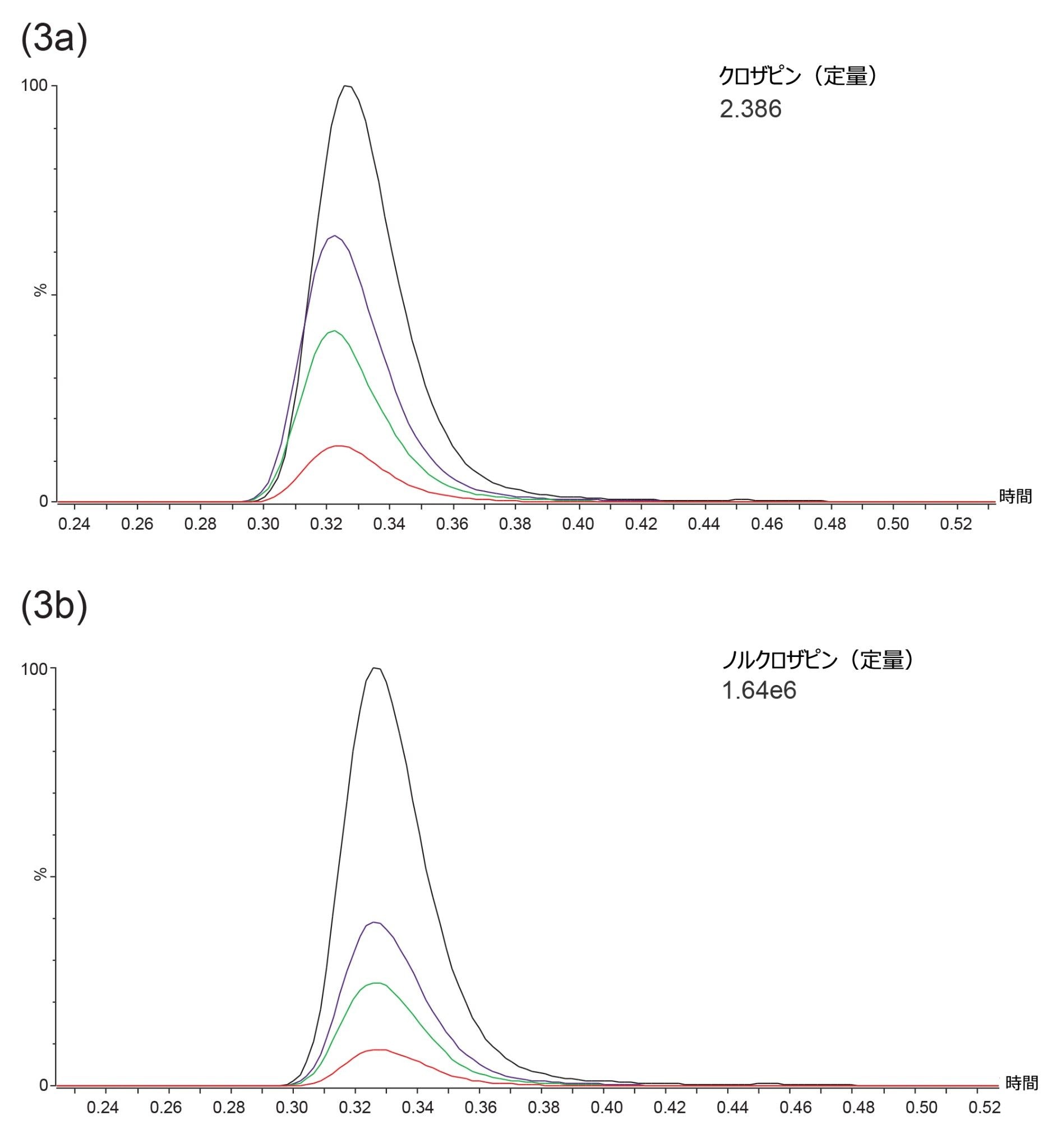 図 3.内部品質管理サンプル中のクロザピン(a)およびノルクロザピン(b)の、代表的なスムージング済み MRM 定量イオントランジションのクロマトグラム。ターゲット/公称管理濃度:150 ng/mL(赤色)、500 ng/mL(緑色)、750 ng/mL(紫色)、2000 ng/mL(黒色)。
図 3.内部品質管理サンプル中のクロザピン(a)およびノルクロザピン(b)の、代表的なスムージング済み MRM 定量イオントランジションのクロマトグラム。ターゲット/公称管理濃度:150 ng/mL(赤色)、500 ng/mL(緑色)、750 ng/mL(紫色)、2000 ng/mL(黒色)。
分析種と内部標準のピーク面積レスポンス比は、同等濃度の溶媒標準試料からのものと比較して、血漿から抽出されたクロザピンでは 98.2 ~ 101.7%(平均 100.3%、n = 6)で、ノルクロザピンでは 101.9 ~ 105.5%(平均 104.1%、n = 6)でした。残留サンプルマトリックスによるレスポンス比の変化は 5.5% 以下でした。この値は最大許容誤差である 10% バイアス以下を下回っており、2H4-クロザピンが、クロザピンだけでなく、ノルクロザピンの定量用の内部標準としても代用できることを示しています。
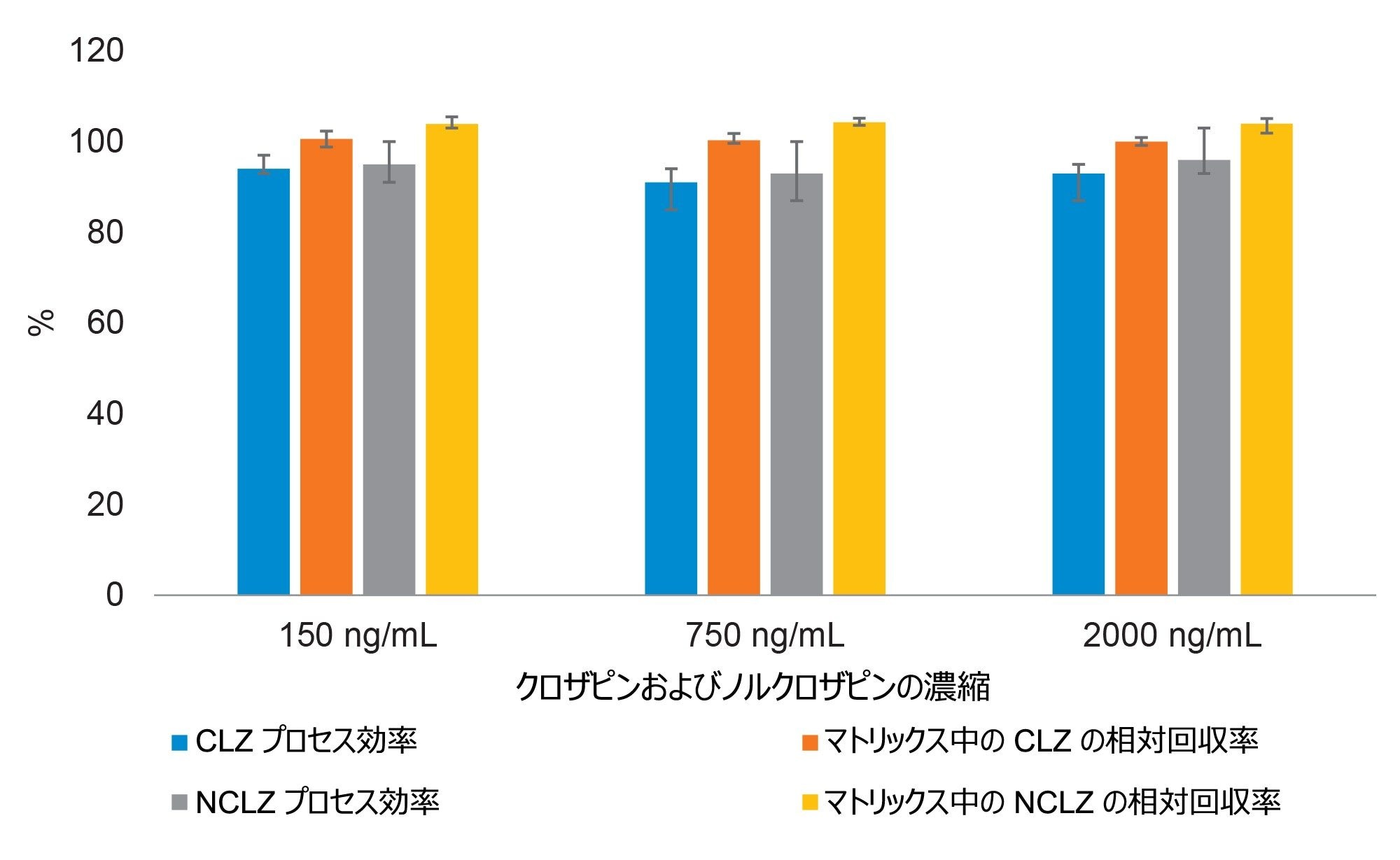 図 4.6 人の健康な成人献血者からの血漿サンプルを使用し、各レベルで 3 回測定して得られた、クロザピンおよびノルクロザピンの平均プロセス効率およびマトリックス効果。プロセス効率は、抽出前スパイクサンプルの分析種対内部標準のピーク面積レスポンス比で、抽出後スパイクサンプルのレスポンスに対して標準化した値です。マトリックス中での相対回収率は、抽出後スパイクサンプルのレスポンスで、同じ濃度のマトリックスを含まない溶媒標準試料に対して標準化した値です(エラーバーは結果の範囲を表します)。
図 4.6 人の健康な成人献血者からの血漿サンプルを使用し、各レベルで 3 回測定して得られた、クロザピンおよびノルクロザピンの平均プロセス効率およびマトリックス効果。プロセス効率は、抽出前スパイクサンプルの分析種対内部標準のピーク面積レスポンス比で、抽出後スパイクサンプルのレスポンスに対して標準化した値です。マトリックス中での相対回収率は、抽出後スパイクサンプルのレスポンスで、同じ濃度のマトリックスを含まない溶媒標準試料に対して標準化した値です(エラーバーは結果の範囲を表します)。
LGC Axio EQA サンプル(TM230 ~ TM240、n=11)の分析。クロザピンの濃度範囲 118 ~ 2210 ng/mL、ノルクロザピンの濃度範囲 94 ~ 2300 ng/mL をカバーしており、良好な分析性能を示しました。平均(範囲)z スコアは、クロザピンについて 0.48(-0.35 ~ 0.97)、ノルクロザピンについて 0.33(-0.21 ~ 0.67)であり、このスキームで十分とされる性能基準は z スコア 2.00 以下でした。ブランド-アルトマン分析では、両方の医薬品について、開発した分析法と割り当て値の間の平均差は 5% 未満でした。
開発した(被験)LC-MS 分析法と比較対象の LC-MS 分析法の関係は、被験クロザピン(ng/mL) = -10.0 + 0.996 × 比較対象クロザピン(ng/mL)、被験ノルクロザピン(ng/mL)= -8.5 + 0.996 × 比較対象ノルクロザピン(ng/mL)であり、n = 76 のサンプルは、クロザピンでは 0 ~ 1003 ng/mL、ノルクロザピンでは 0 ~ 539 ng/mL の範囲でした。いずれの分析種でも比例バイアスの兆候はなく、傾き(95% 信頼区間)は、クロザピンでは 0.996(0.977 ~ 1.016)、ノルクロザピンでは 0.996(0.972 ~ 1.018)でした。被験分析法では、クロザピンについて 10 ng/mL(5 ~ 18)、ノルクロザピンについて 9 ng/mL(5 ~ 13 ng/mL)の固定負バイアス(95% 信頼区間)が見られました。これはおそらく、分析法のキャリブレーションとサンプルの安定性の違いによって説明されます。
MRM イオントランジションの分析的選択性は、候補干渉物質の注入溶媒標準試料のクロマトグラムを調べることで確認されました。説明した LC 分析法および MS/MS 分析法を使用して、30%(v/v)メタノール(水系)中の 20 ng/mL アミトリプチリン、クロミプラミン、ドキセピン、イミプラミン、マプロチリン、ノルクロミプラミン、デシプラミン、ノルドキセピン、ノルマプロチリン、ノルトリミプラミン、ノルトリプタリン、プロトリプチリン、トリミプラミン、25 ng/mL テマゼパム、トリアゾラム、ミダゾラム、オキサゼパム、クロナゼパム、フルラゼパム、クロルジアゼパム、クロバザム、ジアゼパム、ロラゼパム、100 ng/mL シタロプラム、デスメチルフルオキセチン、デュロキセチン、フルオキセチン、O-デスメチルベンラファキシン、セルトラリン、ベンラファキシン、50 ng/mL ミルタザピン、300 ng/mL トラゾドンを注入した場合、クロザピンまたはノルクロザピンの保持時間(0.32 分)の周囲にピークやクロマトグラムのベースライン干渉は認められませんでした。ノルクロザピンおよびクロザピンのチャンネルにそれぞれ、最大許容ピーク面積 35 カウントおよび 205 カウントを用いた場合(測定区間の下限の 25% と予想されるおおよそのピーク面積)、干渉ピークは見られませんでした(データは示していません)。このことから、被験化合物が、ターゲット分析種の波形解析およびその後の定量を妨害する可能性は低いと考えられます。
さらに、クロザピンおよびノルクロザピンの対照サンプルに一部の内因性化合物を添加して、クロザピンおよびノルクロザピンの全体的な回収率に対する潜在的干渉を評価しました。クロザピン/ノルクロザピンの公称濃度は 150 ng/mL(低)および 2000 ng/mL(高)でした。干渉物質候補の存在下でも回収率は最大 5.4% しか低下しませんでした(対照の各セットで n = 3)。クロザピンおよびノルクロザピンの定量が、試験した内因性化合物(アルブミン、コレステロール、トリグリセリド、ビリルビン、尿酸、クレアチニン)の濃度変化の影響を受ける可能性は低いと考えられます。
結論
血漿サンプル中のクロザピンおよびノルクロザピンの迅速定量のための、分析性能特性が優れたシンプルな臨床研究分析法が開発されました。
720008286JA、2024 年 3 月