効率の向上:ACQUITY™ QDa™ II 質量検出器を使用した、医薬品賦形剤中の亜硝酸塩含量の迅速かつ信頼性の高い分析
要約
アプリケーションのメリット
- 29 日間にわたる包括的な分析を通して、ACQUITY™ QDa™ II 質量検出器の頑健な機能を実証し、代表的な医薬品賦形剤中の亜硝酸塩含有量のルーチン測定に使用できることを示す
- メンテナンスの間隔についてオペレーターに実用的な情報を提供し、装置のダウンタイムを記録するなど、装置の使いやすさに焦点を当て、サンプルスループットの高いラボへの適合性を評価する
- ルーチンの分析試験の状況において、亜硝酸塩を µg/g レベルで定量する分析法の精度を評価し、適用したワークフローにおけるシステムの信頼性を評価する
- Empower™ クロマトグラフィーデータシステム(CDS)ソフトウェアを採用して、コンプライアンスに対応した包括的な結果のレポート作成を確実に行います
はじめに
近年、医薬品中の N-ニトロソアミンの存在は、医薬品業界内で急速に懸念事項として認識が進んでいます。安全とみなされるレベルを超えるレベルのこのような不純物に対する長期間の曝露により、患者に発がん性および変異原性のリスクを与える可能性があるという考えから、世界中の規制当局が、複数の薬物クラスにおける N-ニトロソアミン汚染に関する安全性の懸念を提起しています1,2。 その結果、リスク低減およびこれらの化合物の存在の管理が、医薬品開発および品質評価の不可欠な部分になりました。
亜硝酸塩は、N-ニトロソアミンの生成における最も重要なリスク要因です3。 亜硝酸塩が通常酸性条件下で第 2 級アミンまたは第 3 級アミンと反応すると、反応性の NO+ キャリア分子種が生成され、これがニトロソ化剤として働きます。このニトロソ化剤と反応しやすいアミンが特定の条件下において広範に相互作用すると、これが最終的な医薬品製剤内に存在すると考えられるすべての N-ニトロソアミンのレベルに大きく影響します。結果として、このような反応の単一の基礎前駆体としての総亜硝酸塩含量をスクリーニングすることが、リスク評価をサポートする簡単で実用的なアプローチになると考えられます。これにより、N-ニトロソアミン生成のリスクを効率的に軽減することができます。このことにより、感度要件があるために通常はより困難になる従来のニトロソアミン分析の実行要件を低減することができます。
亜硝酸塩汚染は通常、合成中に、溶媒や試薬だけでなく、原材料、中間体、賦形剤からも持ち込まれます。一方、賦形剤中に不純物として存在する亜硝酸塩は通常、ppm レベルで検出され、ニトロソ化剤の主なソースになっています4,5。 亜硝酸塩レベルのばらつきは、賦形剤の種類だけでなく、賦形剤の供給メーカー間でもよく認められます。同じ賦形剤でも異なるベンダーから購入した場合、おそらく製造工程の違いにより、全く異なるレベルの亜硝酸塩濃度を示すことがあります6。
このアプローチではサンプルマトリックスがさまざまであり、試験するサンプル量が増加するため、医薬品メーカーが、供給業者、賦形剤、さらには使用するバッチに関係なく、使用する原材料に汚染がないと確信することができるように、賦形剤中の亜硝酸塩濃度を評価するルーチン分析法の必要性が浮き彫りになっています。このような分析法により、汚染された原材料のフラグ付けが可能になり、必要に応じてさらなる確認試験に使用できます。このリスク評価は迅速で費用対効果が高いことが必要です。また、亜硝酸塩の評価では低コストの分析法が一般的に使用されていますが、これらは多くの場合時間がかかり、感度が低く、干渉物質の影響を受けるという問題があります7。
液体クロマトグラフィーと組み合わせたシングル四重極質量分析(LC-MS)により、微量レベルの亜硝酸の分析に必要な感度と選択性が得られますが、最終的にはいずれのソリューションも、ハイスループットのルーチンワークフローに必要な頑健性と信頼性が求められます。また、1 サンプルあたりの分析が低コストで行えることが求められます8。
以下に説明する実験では、シングル四重極質量検出器に接続した UHPLC を使用して、29 日間の分析期間にわたる医薬品賦形剤の長期分析におけるこの分析設定の適合性を評価しました。この研究の目的は、ACQUITY QDa II 質量検出器の経時的な信頼性と頑健な性能を実証することでした。この提案された分析法はJireš および Douša による UPLC™ 分析法から採用されたもので、その利点として分析速度、感度、検出の選択性が挙げられます8。 この分析のため、既存の分析法を、医薬品賦形剤の効果的な分析に必要な性能、頑健性、信頼性を備えた、費用対効果の高い代替システムである ACQUITY Arc™ システムに移管しました。
実験方法
市販の賦形剤サンプルを購入し、使用するまで室温で保管しました。ソルビトール、ラクトース一水和物、トウモロコシデンプン、マルトデキストリン、炭酸カルシウムを、一般的な医薬品賦形剤の代表として選択しました。購入した化学薬品と試薬はすべて分析グレードの品質であり、UPLC グレードの溶媒をすべてのサンプル、溶液、移動相の調製に使用しました。
図 1 に示すように、Jireš および Douša のプロトコルから改変したプロトコルに従ってサンプルを抽出しました。ここで、粉末状の賦形剤は 2,3-ジアミノナフタルレン(DAN)誘導体化メソッドで得たものです8。 このメソッドの原理は、亜硝酸イオンと DAN から 1H-ナフト(2,3-d)トリアゾール(NAT)が生成する反応で、これを質量検出器を用いて分離および検出することができます。次に、NAT 化合物を、サンプル中の総亜硝酸塩生成の尺度として定量します。
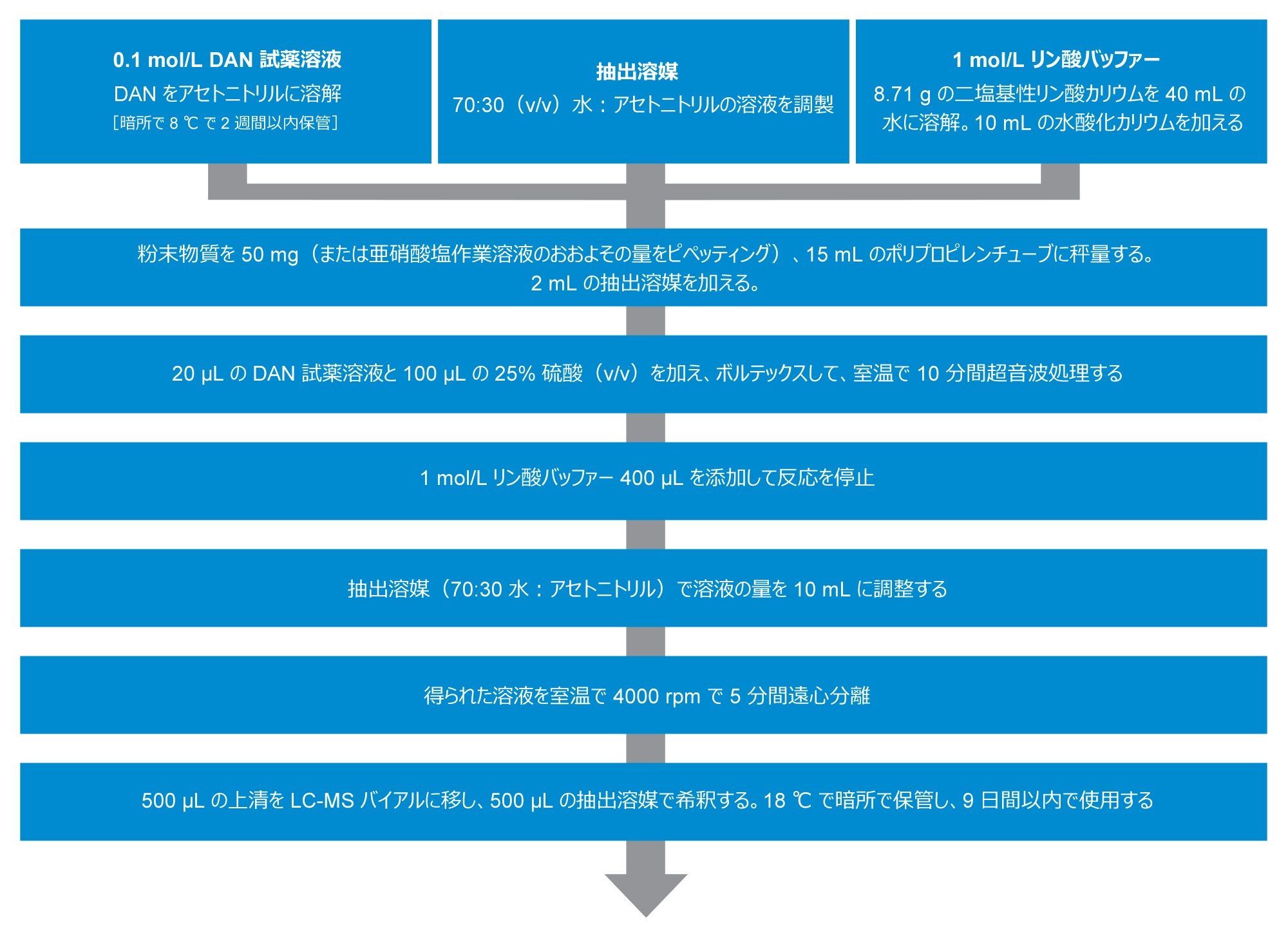 図 1.Jireš および Douša(2022)によるサンプル前処理メソッドを使用しました。
図 1.Jireš および Douša(2022)によるサンプル前処理メソッドを使用しました。
検量線の作成には、亜硝酸スパイク溶液(1 µg/mL)を使用しました。この溶液を、空のポリプロピレン製チューブにさまざまな濃度(0.5、5.0、12.5 ng/mL、賦形剤サンプル中 0.2、2.0、5.0 µg/mL に対応)になるように添加してから、サンプル前処理手順に従って前処理を行いました。次に、スパイクを行っていない賦形剤サンプルを検量線に照らして定量しました。このサンプル前処理手順に特有の亜硝酸塩の相互作用のベースラインレベルを評価するために、プロセスブランク(手順上のブランク)サンプルも調製しました。100 µL の抽出溶媒をプロセスブランクのスパイクの代わりに使用しました。
回収率は、各種の賦形剤 50 mg に対応する量の溶液を各スパイクレベル(0.5、5.0、12.5 ng/mL)になるようにスパイクすることで評価しました。各バッチのすべてのサンプルスパイクは 3 回繰り返しで行いました。
頑健性試験の目的でシステムの安定性を評価およびモニターするために、アセトアミノフェンとカフェインを含む装置 QC 標準試料を 2 種類の濃度レベル(0.5 µg/mL および 5 µg/mL)になるように抽出溶媒中に調製し、分析の過程全体を通して分析しました。
記載したサンプル前処理手法を使用してサンプルを 4 連続のサンプルバッチの期間にわたって調製しました。つまり、サンプル前処理手順全体を計 4 回繰り返しました。選択イオンレコーディング(SIR)は、NAT の場合は m/z 170.1 のターゲットイオンを [M+H]+ として、アセトアミノフェンの場合は m/z 152.1 のターゲットイオンを [M+H]+ として、カフェインの場合は m/z 195.1 のターゲットイオンを [M+H]+ として取り込みました。
分析は、ACQUITY QDa II 質量検出器に接続された ACQUITY Arc システムを使用して行いました。Jireš および Douša の分析法で指定されている UPLC 条件を採用し、システムの圧力限界範囲内に収まった状態で UHPLC システムに確実に移管できるように、カラムカリキュレーター 2.0 ソフトウェア(667005222)を使用しました8。 これにより、システム全体として期待される性能と感度を損なうことなく、仕様が縮小した LC システムを使用することができ、費用対効果の高い分析が確保されました。
LC-MS の実験条件
|
LC システム: |
ACQUITY Arc(FTN-R サンプルマネージャー搭載) |
|
検出: |
ACQUITY QDa II 質量検出器 |
|
カラム: |
ACQUITY UPLC HSS™ T3 カラム、100 Å、1.8 µm、2.1 × 100 mm(製品番号:186003539) |
|
カラム温度: |
45 ℃ |
|
サンプル温度: |
18 °C |
|
注入量: |
5 µL |
|
流速: |
0.4 mL/分 |
|
実行時間: |
6 分 |
|
移動相 A: |
0.1% ギ酸水溶液 |
|
移動相 B: |
アセトニトリル |
|
バイアル: |
透明ガラス 12 × 32 mm、スクリューネックバイアル、100 個入り(製品番号:186000273) |
|
イオン化: |
ポジティブエレクトロスプレー(ES+) |
|
キャピラリー電圧: |
0.8 kV |
|
脱溶媒温度: |
400 度 |
|
イオン源温度: |
120 ℃ |
|
取り込みモード: |
SIR |
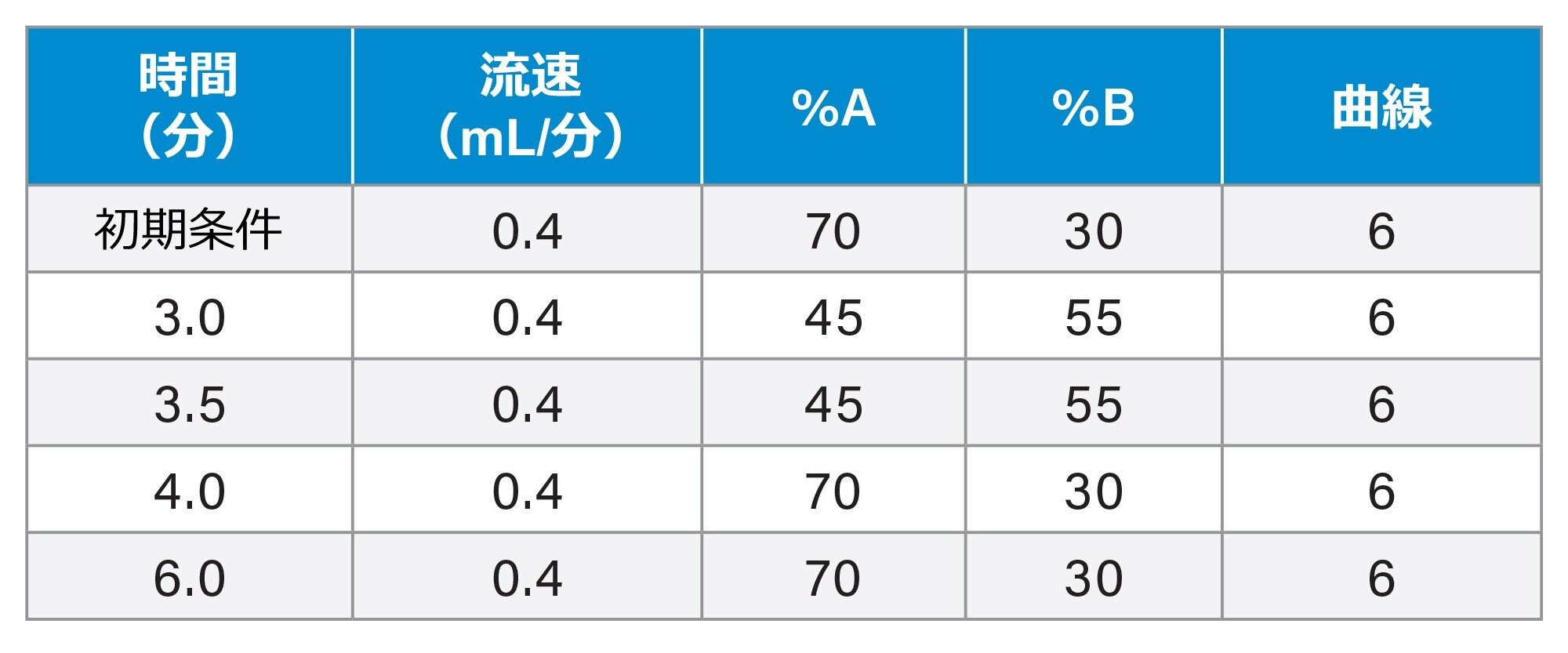
LC グラジエント
ソフトウェア
|
データ取り込み、解析、レポート作成: |
Empower™ CDS 3.0 |
結果および考察
分析法の性能
上記の LC-MS 分析法条件を使用して NAT(m/z 170)を定量することで、選択した賦形剤中の亜硝酸塩含有量を迅速に定量することができました。NAT 化合物の保持時間(RT)は約 2.73 分でした。0.5 ~ 12.5 ng/mL(賦形剤サンプル中の亜硝酸塩濃度 0.2 ~ 5.0 µg/g(µg/g = ppm)に対応)の範囲の検量線を使用して、サンプルを定量しました。
この分析法の LOQ は、プロセスブランクに存在する亜硝酸塩によって制限されます。その例を図 2 に示しており、亜硝酸塩標準試料の絶対ピーク面積をプロセスブランクの横に示しています。
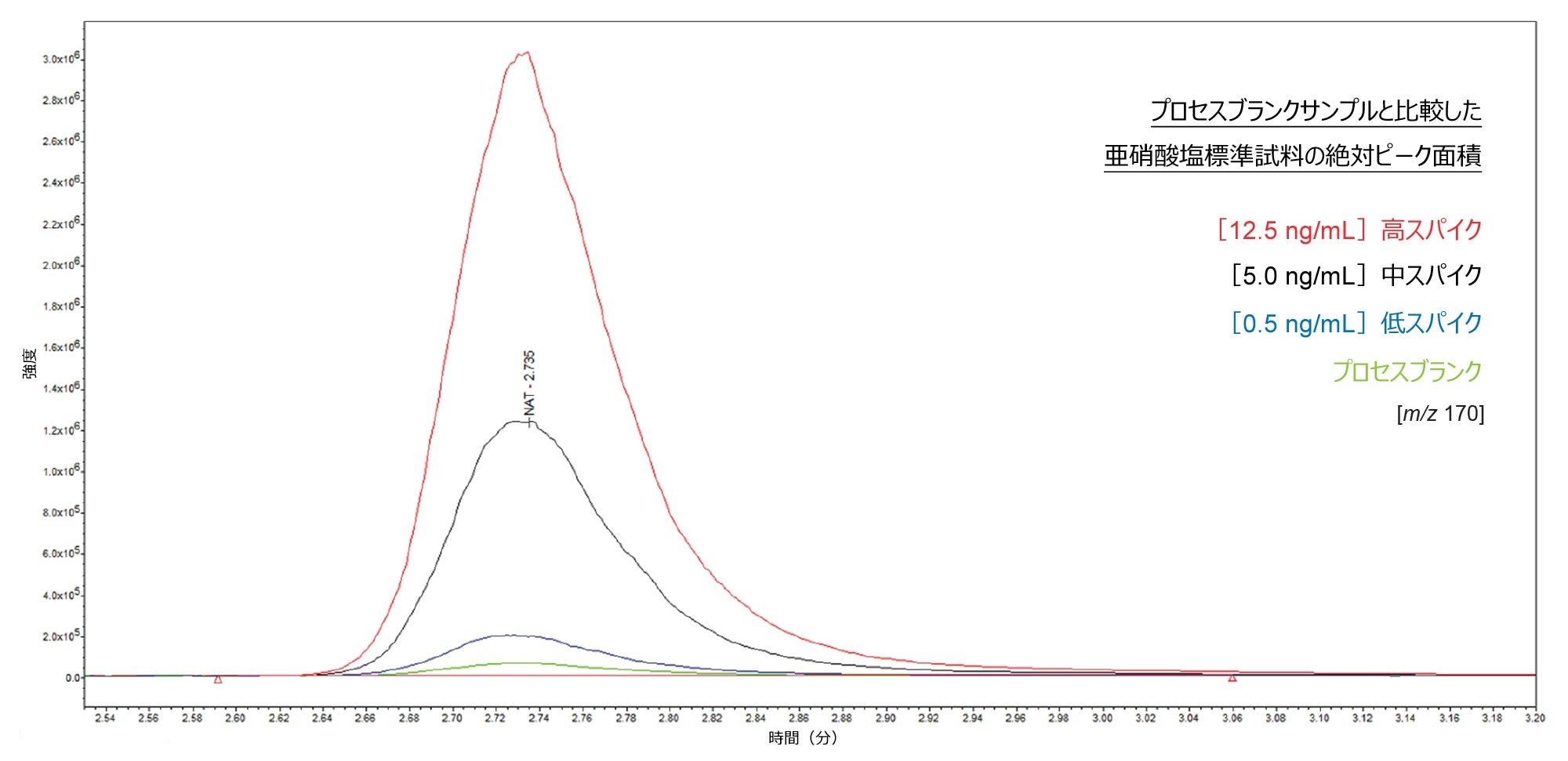 図 2.サンプルシーケンスの所定の繰り返しについて、プロセスブランクに対して重ね描きした亜硝酸塩標準試料の絶対ピーク面積。
図 2.サンプルシーケンスの所定の繰り返しについて、プロセスブランクに対して重ね描きした亜硝酸塩標準試料の絶対ピーク面積。
検量線プロットは補正済み面積を使用して構築しました。ここで、プロセスブランク(サンプルセットごとに n = 6)のピーク面積の平均値を合計し、すべての標準試料および未知試料のピーク面積から減算しました:補正済み面積 = 面積 - (BL?.%..AVE(面積))。このブランク減算の方法を使用すると、調整済み検量線プロットの X 切片がプロセスブランクの代表値(サンプル前処理手法における汚染に由来する各サンプル中に存在する亜硝酸塩のレベル)になります。次に、切片を上回るレスポンスの増加を用いて、後続のサンプル中の亜硝酸塩含有量を定量します。
ハイスループットのワークフローを代表させることを意図した、事前定義された注入シーケンス(181 回の注入)を作成し、148 ~ 190 時間のサンプルバッチにわたって複数回分析しました。ここでの各サンプルバッチ(n = 4)は、スパイク済み賦形剤またはスパイクなしの賦形剤、亜硝酸塩標準試料、プロセスブランク、QC などの新しいサンプルの調製物を意味します。計 29 日間の分析にわたって、サンプル前処理を 4 回繰り返し、注入シーケンスを計 32 回繰り返しました。この分析は、システムの頑健性を実証するように設計したもので、LC の移動相および溶媒のルーチン置き換えを除き、中断することなく連続的に行うことを意図しました。イオン源のクリーニングなどの追加の操作を表 1 に記録しました。
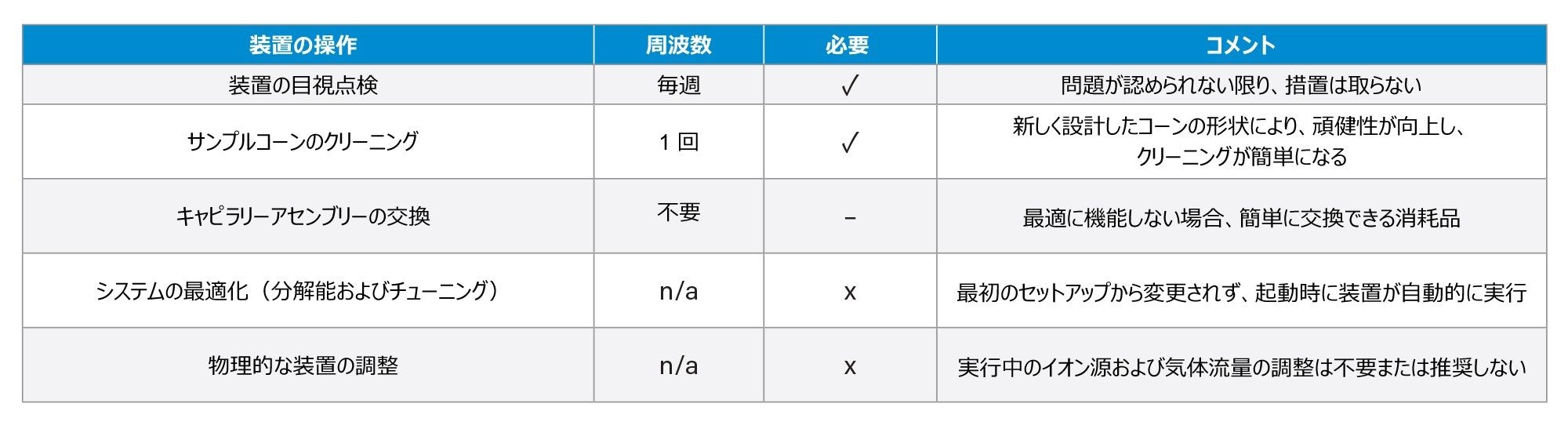 表 1.ACQUITY QDa II 質量検出器での代表的な装置の操作および分析の期間中に行った各作業の頻度。
表 1.ACQUITY QDa II 質量検出器での代表的な装置の操作および分析の期間中に行った各作業の頻度。
分析全体にわたって、事前定義された注入シーケンス(n = 32)の繰り返しごとに、Empower で検量線プロットを作成しました。各注入シーケンスは個別に処理しました。また、注入シーケンスにはキャリブレーション標準試料でブラケット化した賦形剤サンプルが含まれており、これらはすべて 3 回繰り返しで調製したものです。各注入シーケンスの検量線は、サンプルバッチ 2(R2 > 0.97)を除き、すべてのケースにおいて R2 > 0.99 で直線性(1/X)でした。図 3 に、ほぼ 150 時間の分析によって隔てられた、所定のサンプルバッチの最初と最後の検量線プロットの比較を示します。この例では、所定のデータポイントに対応する残差値はいずれも 3.7% を超えていません。
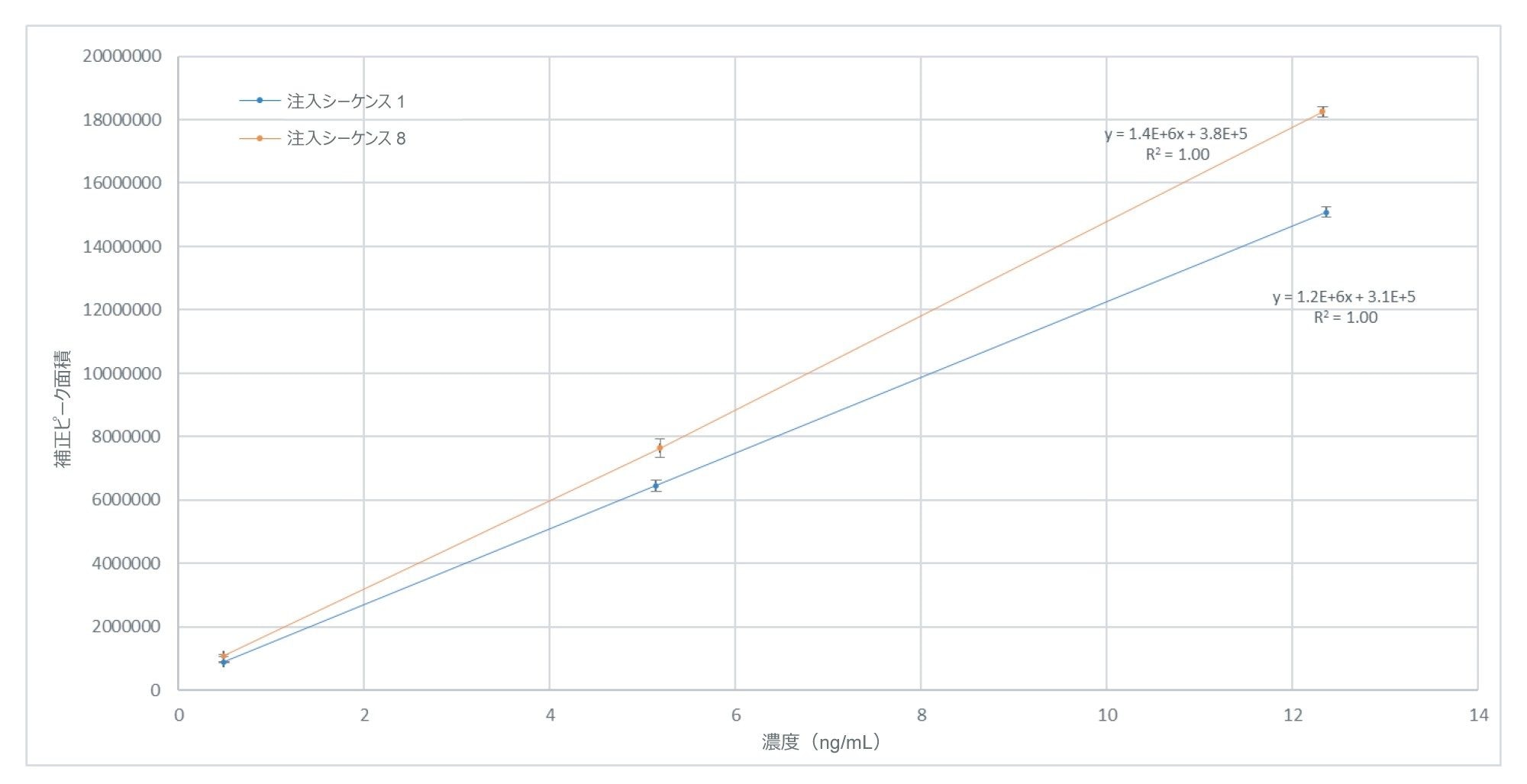 図 3.サンプルバッチの最初と最後にキャリブレーションスタンダードを 3 回繰り返し注入した分析で得られた、0.5 ~ 12.5 ng/mL(原材料中 0.2 ~ 5 µg/g の亜硝酸塩に対応)の濃度範囲にわたる亜硝酸塩(NAT として)のキャリブレーションレスポンス。6 回の注入にわたる各キャリブレーションレベルの平均値とそれぞれの標準偏差。
図 3.サンプルバッチの最初と最後にキャリブレーションスタンダードを 3 回繰り返し注入した分析で得られた、0.5 ~ 12.5 ng/mL(原材料中 0.2 ~ 5 µg/g の亜硝酸塩に対応)の濃度範囲にわたる亜硝酸塩(NAT として)のキャリブレーションレスポンス。6 回の注入にわたる各キャリブレーションレベルの平均値とそれぞれの標準偏差。
最終サンプルバッチは、それまでのサンプルバッチと較べてわずかに長時間(190 時間)分析したため、分析法の経時的安定性を示すのに最も適しています。この研究の目的のため、スパイク済み賦形剤のピーク面積を、既知の同等のスパイクレベルでのピーク面積と比較することにより、回収率を評価しました。この最終サンプルバッチ全体にわたる回収率は、図 4 に示すように、70 ~ 129% の範囲であり、0.5 ng/mL(0.2 µg/g)のトウモロコシデンプンのスパイクが、許容できる回収率の範囲に収まらない唯一のサンプルでした。すべてのサンプルおよびスパイクにわたる合計平均回収率は 94% でした。
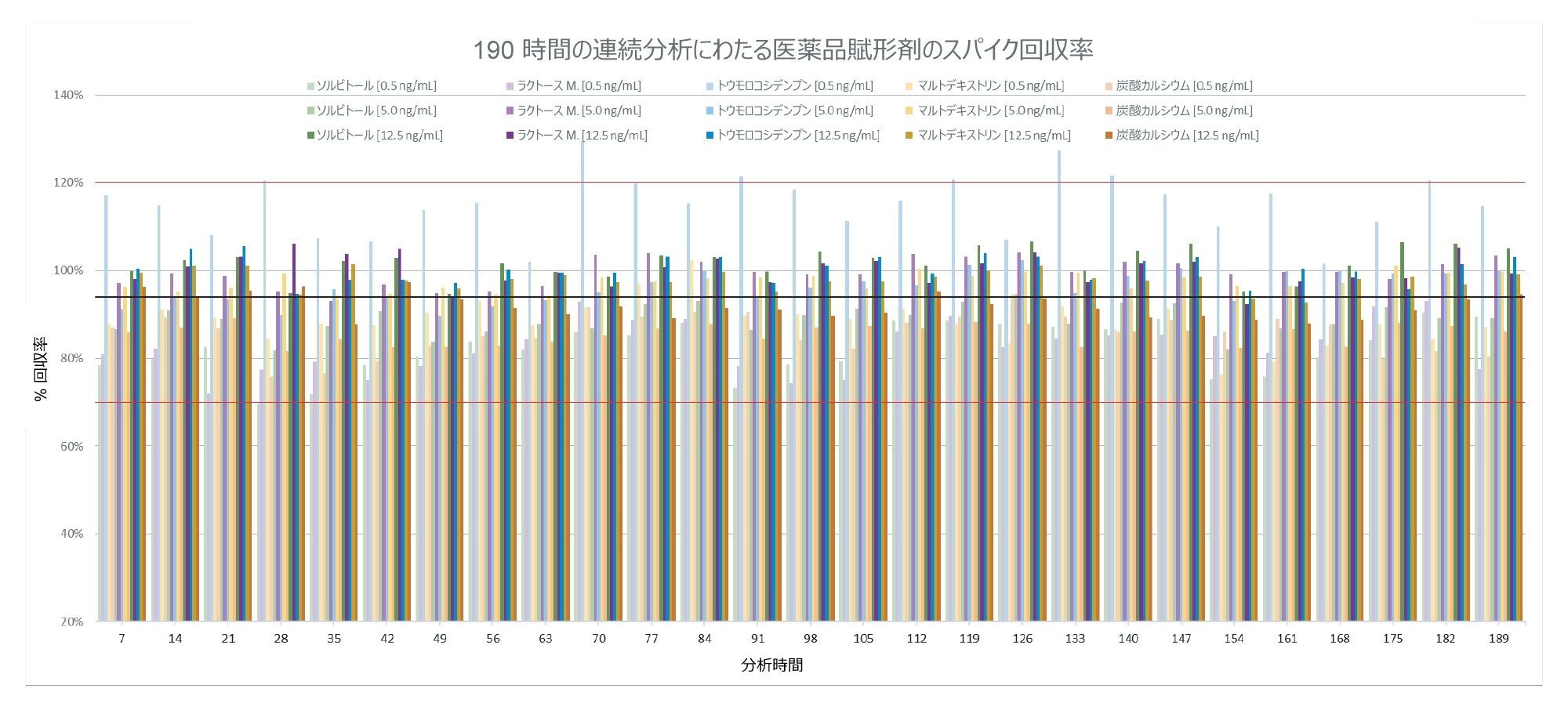 図 4.190 時間の連続取り込みにわたる、スパイク済みた賦形剤サンプルすべてにおける亜硝酸塩の回収率(%)。赤線は許容間隔 70 ~ 120% を示し、黒線は合計平均回収率 94% を示します。
図 4.190 時間の連続取り込みにわたる、スパイク済みた賦形剤サンプルすべてにおける亜硝酸塩の回収率(%)。赤線は許容間隔 70 ~ 120% を示し、黒線は合計平均回収率 94% を示します。
図 5 に、所定のシーケンス内のスパイクなしの各賦形剤の絶対ピーク面積の代表例を示します。重要な点として、プロセスブランクサンプルと賦形剤サンプルは同じサンプル前処理手法を使用して調製されているため、プロセスブランクに本来存在するレベルの汚染は、すべての賦形剤サンプルにわたって存在すると想定されます。図 5 のプロセスブランクに見られる NAT のレベルは 0.041 ng/mL に対応しています。結果として、この図は、トウモロコシデンプンを除いて、分析したすべての賦形剤にわたって亜硝酸塩がほぼ存在しないことを示しています。
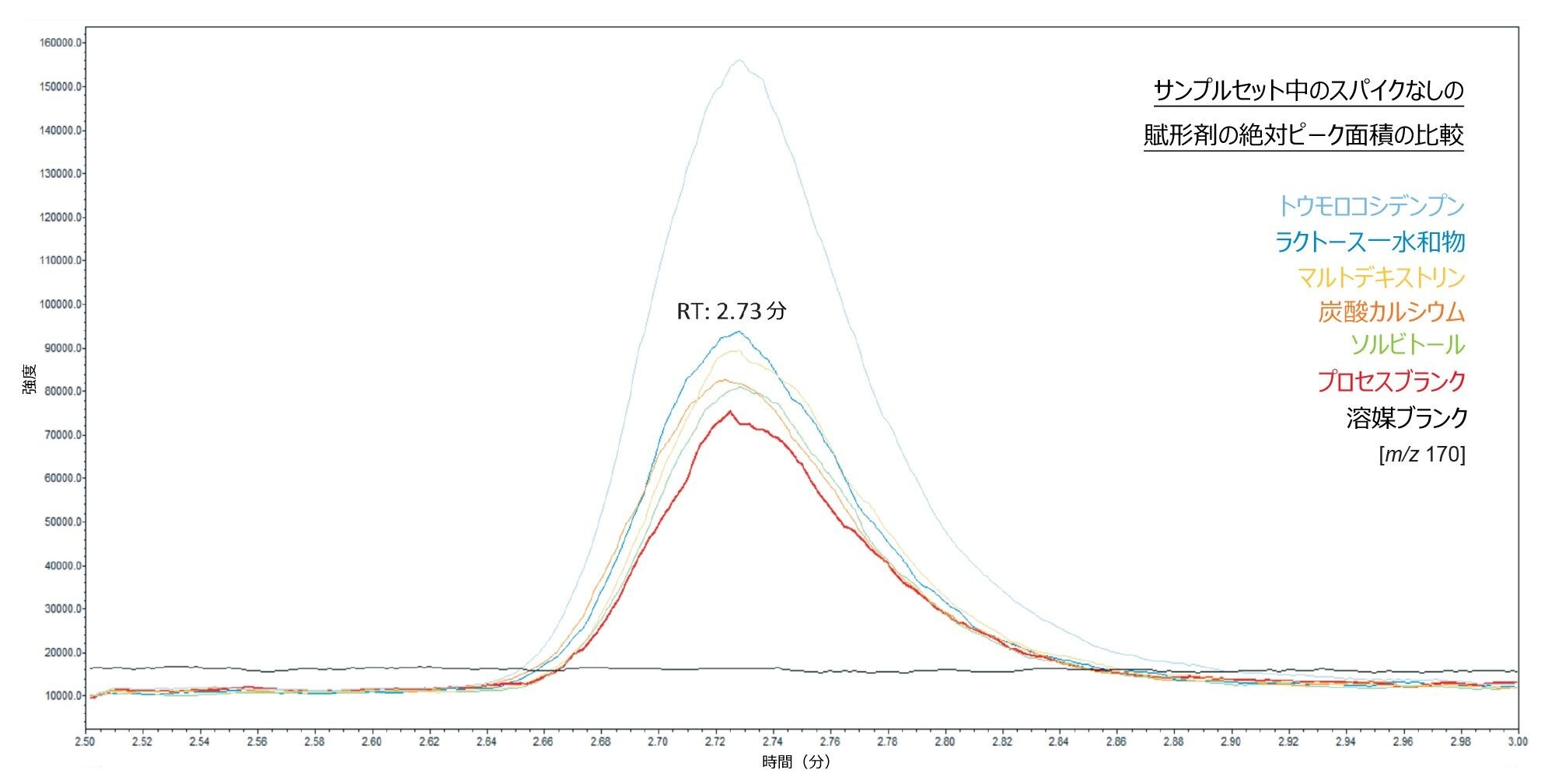 図 5.所定のサンプルシーケンスにおいて、プロセスブランク中の亜硝酸塩の絶対ピーク面積を、スパイクなしの賦形剤サンプルと比較した重ね描き SIR クロマトグラム
図 5.所定のサンプルシーケンスにおいて、プロセスブランク中の亜硝酸塩の絶対ピーク面積を、スパイクなしの賦形剤サンプルと比較した重ね描き SIR クロマトグラム
MS 感度ではなく、プロセスブランク中に亜硝酸塩が存在することが、この分析の制限要因です。この亜硝酸塩の存在は、サンプル前処理時のさまざまなソースに由来する可能性のある汚染に起因します。この分析法は本質的に再現性があり、取り込んだ 4 サンプルバッチにわたって、プロセスブランクの計算値の %RSD は 2.7 ~ 8.5% の範囲ですが、分析法の検出限界(LOD)を下げるために、今後の取り組みではこの汚染をなくすことに重点を置く必要があります。
分析の過程にわたって分析した 5 種類の賦形剤については、トウモロコシデンプンにのみ、さらなる分析が必要と考えられるレベルの亜硝酸塩が含まれることがわかりました。図 6 に、190 時間の分析にわたるスパイクなしのトウモロコシデンプンサンプルの報告濃度を示しています。これによると、27 回の注入にわたる亜硝酸塩含有量の平均値は 0.14 µg/g で、%RSD は 8.7% でした。この値は、賦形剤データベース全体にわたるトウモロコシデンプンの平均値と一致しています6。 点線は、トウモロコシデンプンの公称報告しきい値の推定例で、文献に記載されているように、亜硝酸塩含有量の報告されている平均値の 0.5 倍としてプロットされています。この例では、賦形剤がこのしきい値を超えており、さらなる分析が必要と考えられます。
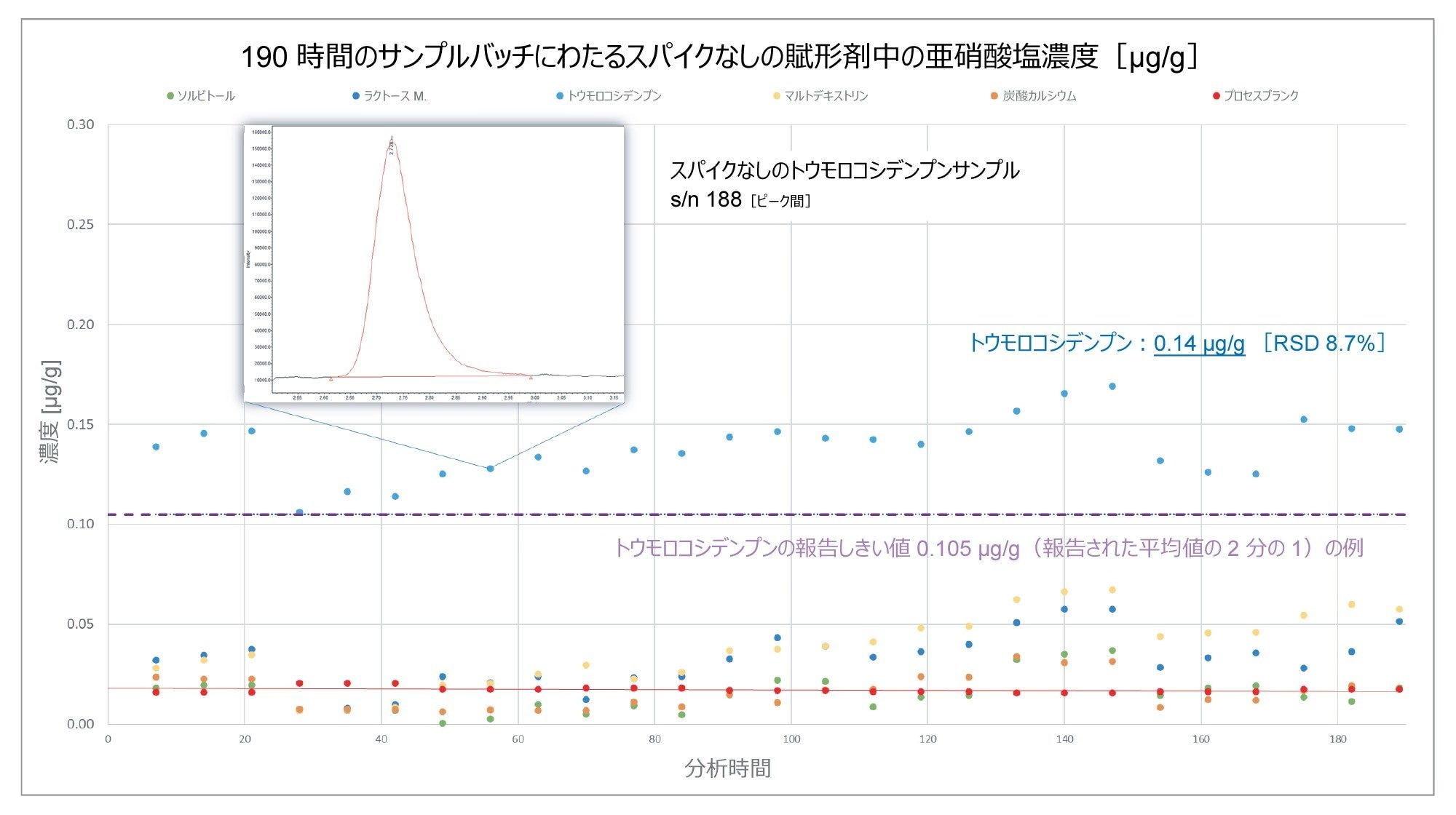 図 6.190 時間の連続取り込みにわたって分析した、スパイクなしの賦形剤サンプル中の亜硝酸塩含有量(µg/g)。トウモロコシデンプンサンプルの亜硝酸塩含有量は、文献で報告されている平均値の 2 分の 1 である 0.105 µg/g を超えていました。27 回の注入にわたるこのサンプル中の亜硝酸塩の測定精度は %RSD 8.7% です。
図 6.190 時間の連続取り込みにわたって分析した、スパイクなしの賦形剤サンプル中の亜硝酸塩含有量(µg/g)。トウモロコシデンプンサンプルの亜硝酸塩含有量は、文献で報告されている平均値の 2 分の 1 である 0.105 µg/g を超えていました。27 回の注入にわたるこのサンプル中の亜硝酸塩の測定精度は %RSD 8.7% です。
これとは対照的に、図 7 では、同じプロセスを用いてラクトース一水和物の報告されたしきい値の例を確認したところ、分析した賦形剤がしきい値 0.27 µg/g を超えていないため、この場合は使用可能と考えられます。
%RSD の大きなばらつきは、賦形剤中の亜硝酸塩含有量がプロセスブランクの亜硝酸塩含有量と近いことを反映しています。2 サンプル間の相対濃度差が非常に小さいことから、賦形剤には検出可能な亜硝酸塩が存在しないと考えられます。したがって、この高い %RSD 値はこの分析法の限界を反映しており、装置性能を反映してはいません。
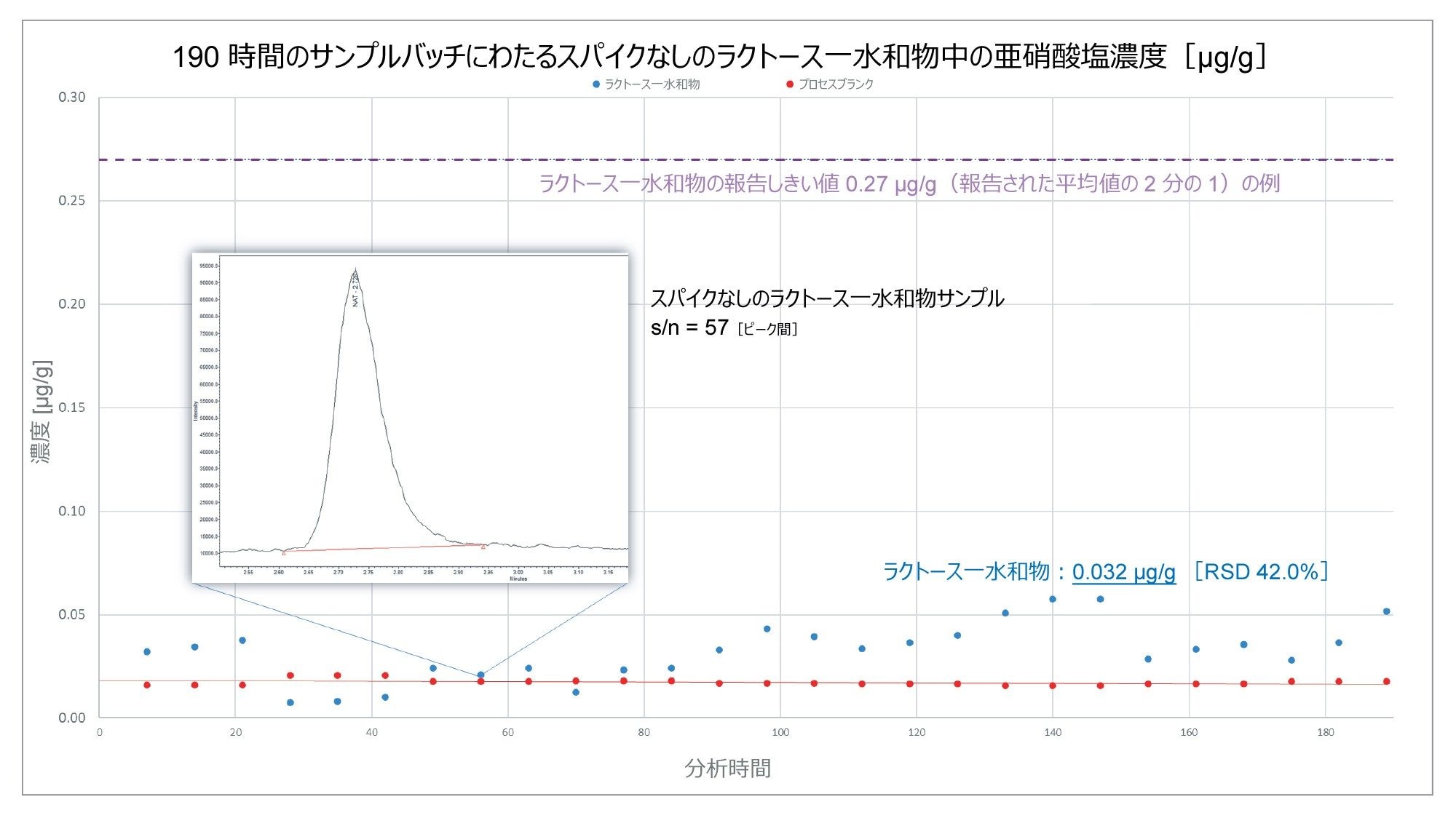 図 7.190 時間の連続分析にわたって分析した、スパイクなしのラクトース一水和物の亜硝酸塩含有量(µg/g)。ラクトース一水和物サンプルの亜硝酸塩含有量は、文献で報告されている平均値の 2 分の 1 である 0.27 µg/g を超えていませんでした。
図 7.190 時間の連続分析にわたって分析した、スパイクなしのラクトース一水和物の亜硝酸塩含有量(µg/g)。ラクトース一水和物サンプルの亜硝酸塩含有量は、文献で報告されている平均値の 2 分の 1 である 0.27 µg/g を超えていませんでした。
所定のサンプルバッチのスパイクなしの他の賦形剤の値も図 6 にプロットしています。また、これらの値は、正確に定量するためのプロセスブランクのしきい値を大きく上回ってはいません。同様に、これらの賦形剤中には亜硝酸塩が存在しないか、あるいは亜硝酸塩のレベルが分析法の検出限界と同程度に低いことを示唆しているため、これらのサンプルはさらなる分析が不要であると考えられます。
図 7 において、スパイクなしのラクトース一水和物サンプルのシグナル/ノイズ比は、分析法の感度が装置の機能ではなくプロセスブランクによる制限を受けていることを示唆しています。システム感度は、(上記の図に挿入されているクロマトグラムの例からわかるように)報告されているしきい値の少し上(図 6)やかなり下(図 7)のレベルを検出するのに十分です。以前に報告されているデータでは、分析法の LOD は 0.0013 ng/mL と示唆されています8。
Empower CDS のレポート機能を使用すると、図 8 からわかるように、サンプルバッチ内の賦形剤の亜硝酸塩濃度を簡単に確認することができます。これにより、ラボ内でのコンプライアンスとデータインテグリティを確保しつつ、迅速で包括的な意思決定を行うことができます。示している例では、スパイクなしのトウモロコシデンプンサンプルは、報告濃度 0.392 ng/mL または 0.157 µg/g と表示されています。
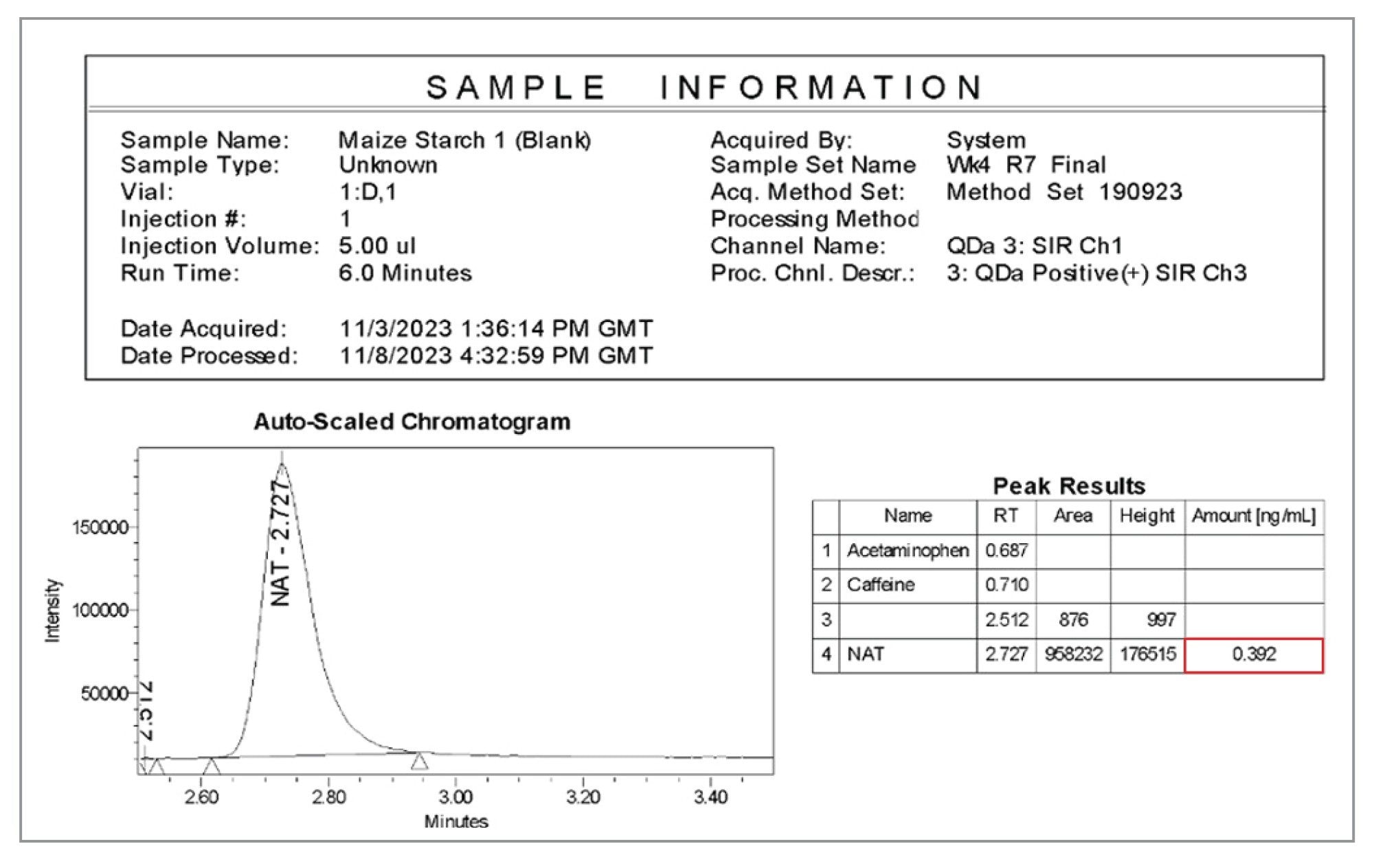 図 8.トウモロコシデンプンサンプルの亜硝酸塩含有量について Empower CDS で生成されたデータレポートの例。
図 8.トウモロコシデンプンサンプルの亜硝酸塩含有量について Empower CDS で生成されたデータレポートの例。
装置の頑健性
分析法の性能だけでなく、装置の全体的な稼働時間と信頼性を評価し、装置の操作およびすべてのメンテナンス手順を分析全体にわたって記録しました。29 日間にわたって、計 5792 回の注入を装置で行い、そのうち 1920 回は賦形剤サンプルでした。分析の概要を図 9 に示します。
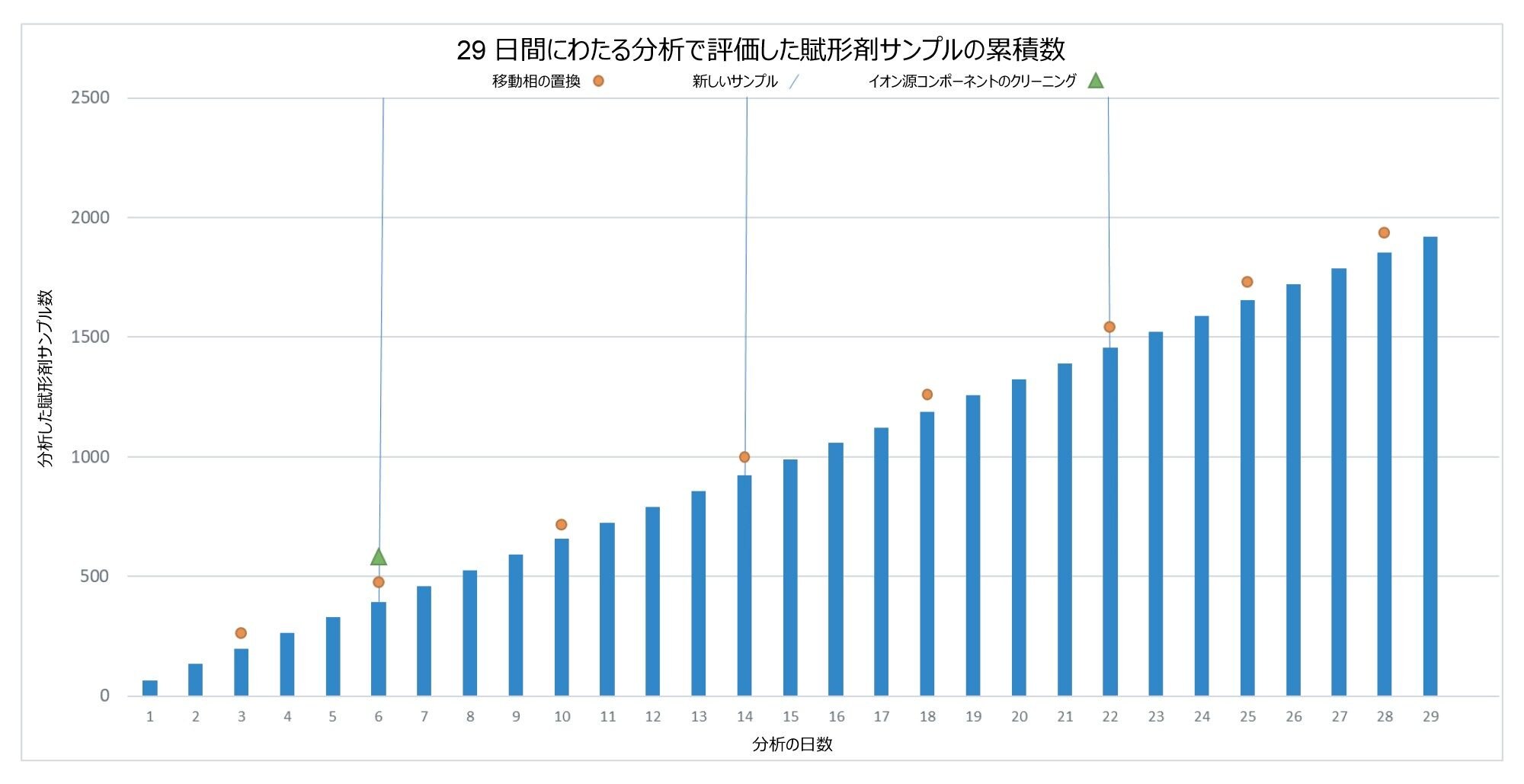 図 9.29 日間にわたって分析した賦形剤サンプルの累積プロット。注釈には移動相の変更、新しいサンプルの導入、必要なクリーニングを示しています。
図 9.29 日間にわたって分析した賦形剤サンプルの累積プロット。注釈には移動相の変更、新しいサンプルの導入、必要なクリーニングを示しています。
29 日間にわたる分析期間中、必要なシステム操作は限定されていました。676 時間のバッチ分析の間、装置に必要なメンテナンスは 2 時間のみで、そのうち 20 分間は真空解除およびクリーニングのプロセスでした。システムは 2 時間以内に分析を再開しました。6 日後にイオン源のクリーニングを行ったにもかかわらず、システムはその後 23 日間完全に動作を続けられたことから、汚染は以前の装置の使用に関連したものである可能性が高いことが示唆されました。追加の形の装置の操作の詳細は、表 1 に記載しています。
QC 注入が許容範囲から逸脱していたため、イオン源のコンポーネントを一度クリーニングしました。装置の分解能、チューニング、キャリブレーションは起動時に自動化されており、分析者の介入は不要でした。
移動相および洗浄溶液は必要に応じて補充しました。いずれの溶媒ブランクでも亜硝酸塩のキャリーオーバーは観察されず(分析全体で n = 1920)、この分析法の使用によりサンプルスループットが向上することが予想されます。
QDa II 質量検出器の頑健性は、このワークフローの実現可能性を評価する上で重要な指標でした。 装置 QC(1 レベルあたり n = 768)を分析の過程にわたって観察して、亜硝酸塩サンプルとは独立して装置の頑健性を評価しました。結果は分析全体にわたって一貫していました。所定のサンプルバッチ内で、所定の QC レベルの %RSD は 9.8% を超えていませんでした。イオン源コンポーネントの洗浄後の 520 時間の連続稼働において、図 10 に示すように、QC 化合物とスパイクレベルの両方で %RSD は 12.5% 未満でした。
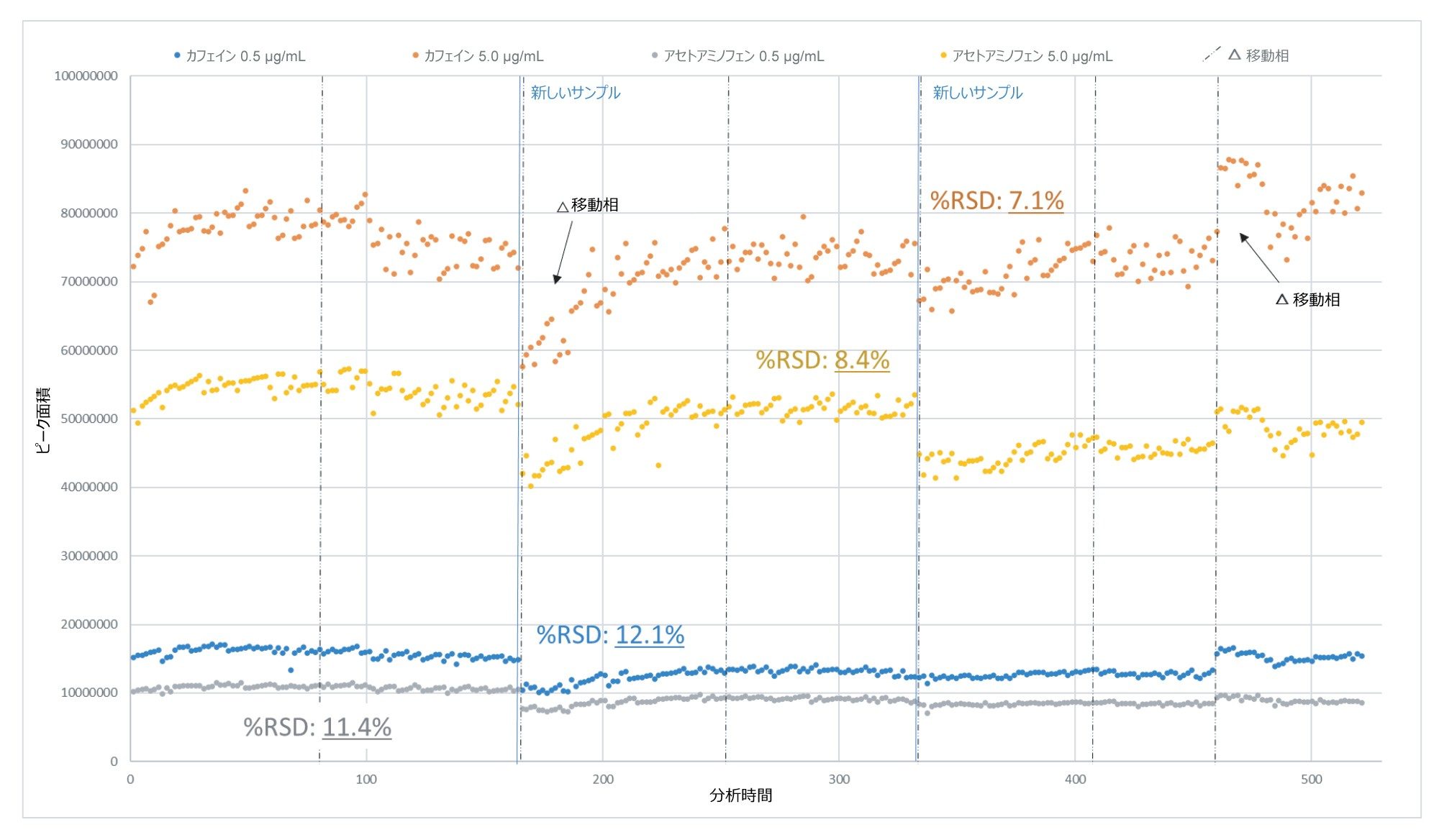 図 10.520 時間の連続分析にわたる QC 成分(カフェインおよびアセトアミノフェン)の絶対ピーク面積(サンプルコーン洗浄前のデータポイントは除外)。
図 10.520 時間の連続分析にわたる QC 成分(カフェインおよびアセトアミノフェン)の絶対ピーク面積(サンプルコーン洗浄前のデータポイントは除外)。
装置の頑健性は、亜硝酸塩標準試料注入の絶対ピーク面積の %RSD を評価することによっても、各サンプルバッチにわたって実証することができます。表 2 からわかるように、%RSD は、分析全体にわたって、所定のサンプルバッチ内で 14.7% を超えていませんでした。
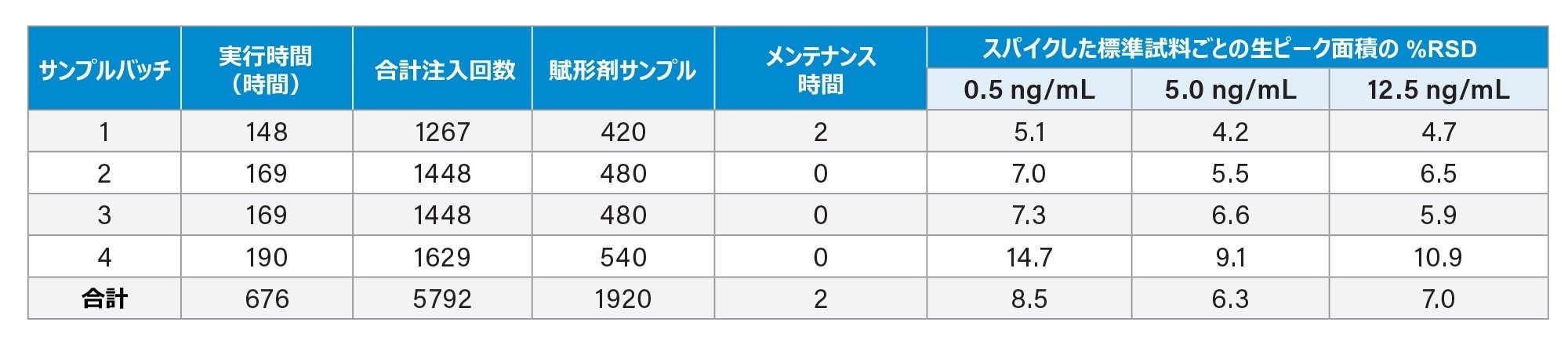 表 2.頑健性試験の過程にわたって分析した注入回数および賦形剤サンプル数の内訳、およびスパイク済み標準試料の絶対ピーク面積の %RSD(すべての所定のスパイクレベルについて n ≥ 42)。
表 2.頑健性試験の過程にわたって分析した注入回数および賦形剤サンプル数の内訳、およびスパイク済み標準試料の絶対ピーク面積の %RSD(すべての所定のスパイクレベルについて n ≥ 42)。
賦形剤へのスパイク全体にわたって報告された濃度は、ほとんど予想と一致していました。スパイク済み賦形剤は、分析全体を通してすべてのスパイクレベルおよび賦形剤の種類にわたって %RSD が 20% 未満でした。例を図 11 に示します。ここでは、分析全体を通して、炭酸カルシウムのすべてのスパイクレベルについて 13.9% 以下の %RSD が示されています。
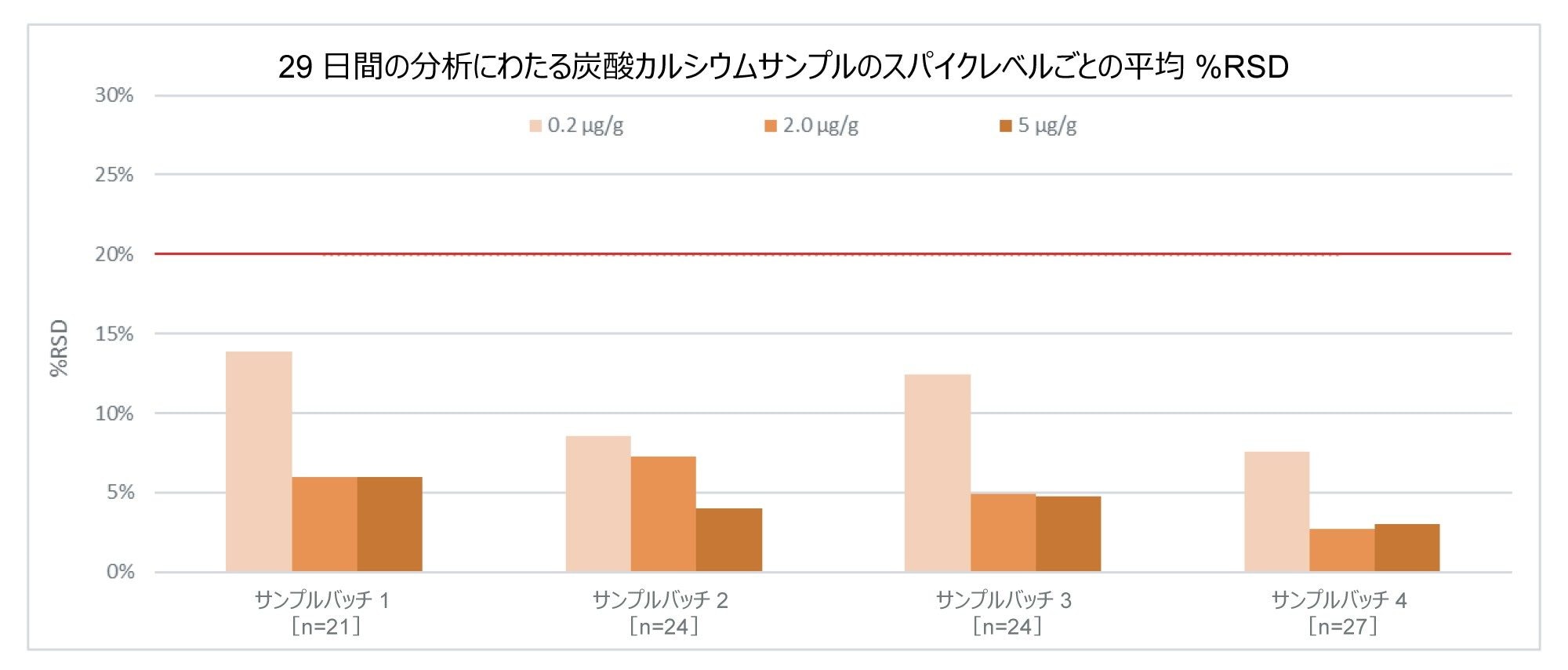 図 11.所定のサンプルバッチにおけるスパイクレベルごとの繰り返し全体にわたる、炭酸カルシウム賦形剤サンプル中 0.2 µg/g、2.0 µg/g、5.0 µg/g 亜硝酸塩スパイクの平均 %RSD。赤線は %RSD のしきい値 20% を示します。
図 11.所定のサンプルバッチにおけるスパイクレベルごとの繰り返し全体にわたる、炭酸カルシウム賦形剤サンプル中 0.2 µg/g、2.0 µg/g、5.0 µg/g 亜硝酸塩スパイクの平均 %RSD。赤線は %RSD のしきい値 20% を示します。
図 12 に、29 日間の分析の過程にわたる各賦形剤サンプルおよびスパイクレベルの平均 %RSD 値を示します。分析全体にわたって、分析した 5 種類の賦形剤のスパイクレベル 0.2、2.0、5.0 µg/g(ppm)にわたる %RSD は、それぞれ 18.5%、11.1%、6.0% を超えておらず、装置性能が時間が経っても頑健であることが実証されました。
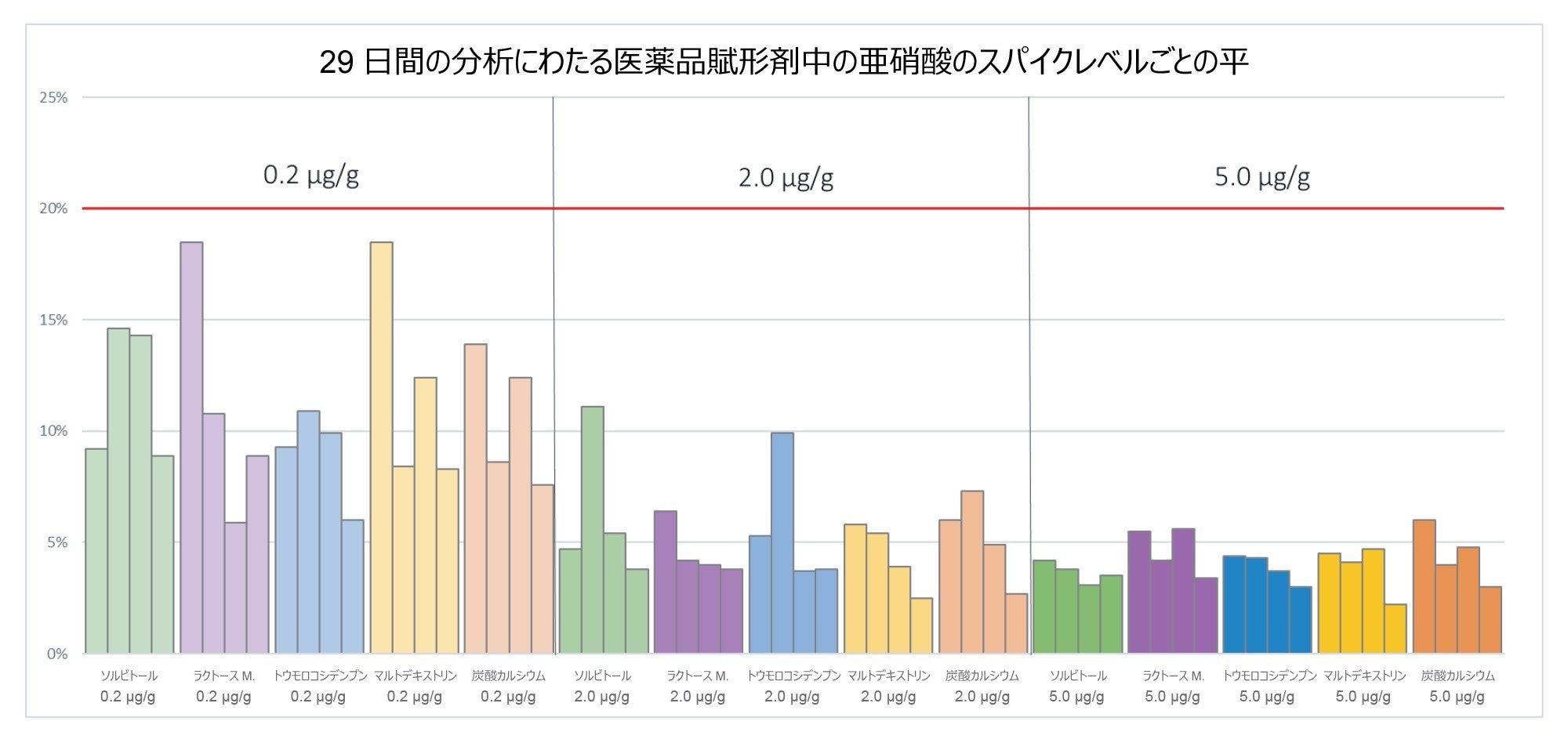 図 12.分析全体にわたるすべての賦形剤スパイクレベルの平均 %RSD。賦形剤スパイクレベルごとの 4 つのデータポイントそれぞれが、所定のサンプルバッチにおけるスパイクレベルごとの平均値を表しています(注:サンプルバッチ 1 の 1 回の繰り返しは、サンプル前処理時のミスにより、0.2 µg/g のマルトデキストリンスパイクレベルから除外しました)。
図 12.分析全体にわたるすべての賦形剤スパイクレベルの平均 %RSD。賦形剤スパイクレベルごとの 4 つのデータポイントそれぞれが、所定のサンプルバッチにおけるスパイクレベルごとの平均値を表しています(注:サンプルバッチ 1 の 1 回の繰り返しは、サンプル前処理時のミスにより、0.2 µg/g のマルトデキストリンスパイクレベルから除外しました)。
結論
- 実施した実験の結果から、ACQUITY QDa II 質量検出器が、医薬品賦形剤中の亜硝酸塩の分析などのルーチンのハイスループットアプリケーションに適した、頑健で信頼性の高い装置であることが分かります
- 装置で 676 時間の分析にわたる約 6000 回の注入を正常に分析することができ、ダウンタイムはわずか 2 時間でした
- 所定のサンプルバッチにおける装置 QC 注入の %RSD は 9.8% を超えておらず、一貫した性能が実証されました
- 520 時間の連続分析を通じて、所定の QC レベルにおける絶対ピーク面積の %RSD は 12.5% 未満に留まっており、分析全体を通してシステムの安定性が優れていることが実証されました
- すべての亜硝酸塩標準試料注入の絶対ピーク面積の %RSD は、分析した所定のサンプルバッチのいずれにおいても一貫して 14.7% 未満でした
- トウモロコシデンプンの賦形剤において、亜硝酸塩レベルが 0.14 µg/g と正常に検出されて、賦形剤データベースに報告されている平均レベルと一致した値になり、分析法の定量的な正確性が示されました。分析において、他の賦形剤には検出可能なレベルの亜硝酸塩が認められませんでした
- ACQUITY Arc UHPLC と ACQUITY QDa II 質量検出器を使用したこの 6 分間の分析法は、医薬品の賦形剤分析における亜硝酸塩の µg/g レベルでの定量に適した、費用対効果の高いアプローチと言えます
参考文献
- Brambilla, G., & Martelli, A. (2007).Genotoxic and carcinogenic risk to humans of drug-nitrite interaction products.In Mutation Research - Reviews in Mutation Research (Vol.635, Issue 1, pp.17–52). https://doi.org/10.1016/j.mrrev.2006.09.003.
- Horne, S., Vera, M. D., Nagavelli, L. R., Sayeed, V. A., Heckman, L., Johnson, D., Berger, D., Yip, Y. Y., Krahn, C. L., Sizukusa, L. O., Rocha, N. F. M., Bream, R. N., Ludwig, J., Keire, D. A., & Condran, G. (2023).Regulatory Experiences with Root Causes and Risk Factors for Nitrosamine Impurities in Pharmaceuticals.In Journal of Pharmaceutical Sciences (Vol.112, Issue 5, pp.1166–1182).Elsevier B.V. https://doi.org/10.1016/j.xphs.2022.12.022.
- Cioc, R. C., Joyce, C., Mayr, M., & Bream, R. N. (2023).Formation of N-Nitrosamine Drug Substance Related Impurities in Medicines: A Regulatory Perspective on Risk Factors and Mitigation Strategies.In Organic Process Research and Development.American Chemical Society. https://doi.org/10.1021/acs.oprd.3c00153.
- Wu, Y., Levons, J., Narang, A. S., Raghavan, K., & Rao, V. M. (2011).Reactive impurities in excipients: Profiling, identification and mitigation of drug-excipient incompatibility.In AAPS PharmSciTech (Vol.12, Issue 4, pp.1248–1263). https://doi.org/10.1208/s12249-011-9677-z.
- Berardi, A., Jaspers, M., & Dickhoff, B. H. J. (2023).Modeling the Impact of Excipients Selection on Nitrosamine Formation towards Risk Mitigation.Pharmaceutics, 15(8). https://doi.org/10.3390/pharmaceutics15082015.
- Boetzel, R., Schlingemann, J., Hickert, S., Korn, C., Kocks, G., Luck, B., Blom, G., Harrison, M., François, M., Allain, L., Wu, Y., & Bousraf, Y. (2023).A Nitrite Excipient Database: A Useful Tool to Support N-Nitrosamine Risk Assessments for Drug Products.Journal of Pharmaceutical Sciences, 112(6), 1615–1624. https://doi.org/10.1016/j.xphs.2022.04.016.
- Wang, Q. H., Yu, L. J., Liu, Y., Lin, L., Lu, R. gang, Zhu, J. ping, He, L., & Lu, Z. L. (2017).Methods for the detection and determination of nitrite and nitrate: A review.In Talanta (Vol.165, pp.709–720).https://doi.org/10.1016/j.talanta.2016.12.044.
- Jireš, J., & Douša, M. (2022).Nitrites as precursors of N-nitrosation in pharmaceutical samples – A trace level analysis. Journal of Pharmaceutical and Biomedical Analysis, 213.https://doi.org/10.1016/j.jpba.2022.114677.
720008197JA、2024 年 2 月