Analysis of 28 EU Regulated and Recommended PFAS in Food via LC-MS/MS – Part 1: Vegetable, Fruit, and Baby Food
Abstract
This study introduces an optimized LC-MS/MS methodology for the comprehensive analysis of Per- and Polyfluoroalkyl Substances (PFAS) in vegetable, fruit, and baby food. The method demonstrates an exceptionally low limit of quantification, reaching 0.0005 µg/kg for some compounds in matrix, while accurately detecting and quantifying the PFAS compounds listed in the Commission Recommendation (EU) 2022/1431. The combination of enhanced sensitivity of the Xevo™ TQ Absolute MS System, with the increased clean-up efficacy of a prototype bilayer dual-phase GCB/WAX SPE cartridge gave excellent recoveries, between 87 and 116% for the mandatory PFAS, and between 65 and 131% for the majority of the recommended compounds along with repeatability (RSDr) ≤10%.
Benefits
- Sensitive quantitative method for the analysis of mandatory, recommended, and considered PFAS in the EU Commission recommendation allowed for all PFAS to be incorporated into a single method meeting requirements
- Extremely low limits of quantification (down to 0.0005 µg/kg) in vegetable, fruit, and baby food allowing to surpass the criteria set by the EURL POPs guidelines
- Excellent method recoveries and repeatability, which complies with the acceptance criteria in the guidelines
- Good recovery for FOSA using a new bilayer dual-phase SPE cartridge SPE cartridge containing GCB and WAX for sample clean-up, allowing to effectively clean-up neutral PFAS
- Enhanced time effectiveness compared to prior UPLC™ methods, resulting in a 50% reduction in sample analysis time, while ensuring baseline separation between linear and branched PFAS compounds and potential interferences
Introduction
Per- and Polyfluoroalkyl Substances (PFAS) constitute a class of synthetic compounds widely used in various industrial and commercial applications, known for their persistence and potential adverse health effects. Regulatory bodies have increasingly prioritized stringent monitoring and control of PFAS levels in food due to their presence in agricultural environments and food chains, posing potential health risks to consumers.
In the European Union (EU), Commission Regulation (EU) 2022/2388, amending Regulation (EC) No 1881/2006, applying from 1 January 2023, sets individual maximum levels for PFOS, PFOA, PFNA, and PFHxS, together with a maximum level for the sum of those PFAS, in foods of animal origin.1 Commission Recommendation (EU) 2022/1431, in force from September 2022, recommends Member States should test for the presence of the same four PFAS during the years 2022, 2023, 2024, and 2025 in a wider range of foodstuffs than covered in 2022/2388, and also suggests the monitoring for a larger list of PFAS in various foodstuffs.2
Commission Implementing Regulation (EU) 2022/1428 (CIR), in force from September 2022, provides methods of sampling and analysis for the control of PFAS in certain foodstuffs, it also provides acceptance criteria for validation of methods and information on reporting and interpretation of results.3 Furthermore, in 2022, the EURL POPs released a guidance document for PFAS methods, which provides information on expected method performance and limits of quantification.4,5 AOAC Standard Method Performance Requirements (SMPRs®) describe the minimum recommended performance characteristics to be used during the evaluation of a method. The evaluation may be an on-site verification, a single laboratory validation, or a multi-site collaborative study. AOAC SMPRs can be used as acceptance criteria for verification in scenarios beyond Commission Implementing Regulation (EU) 2022/1428. AOAC SMPR 2023.003 for PFAS in a variety of foodstuffs has been published.6
Part 1 of this study focuses on the development of a sensitive LC-MS/MS method tailored for the detection and quantification of ultra-trace levels of PFAS in vegetable, fruit, and baby food to address the need for reliable analytical methods and to meet the criteria set in the recent guidelines. Part 2 will focus on the determination of PFAS in animal products.
Experimental
Standards and Solutions
All standards were purchased from Wellington Laboratories. The following standards were used to prepare stock solutions:
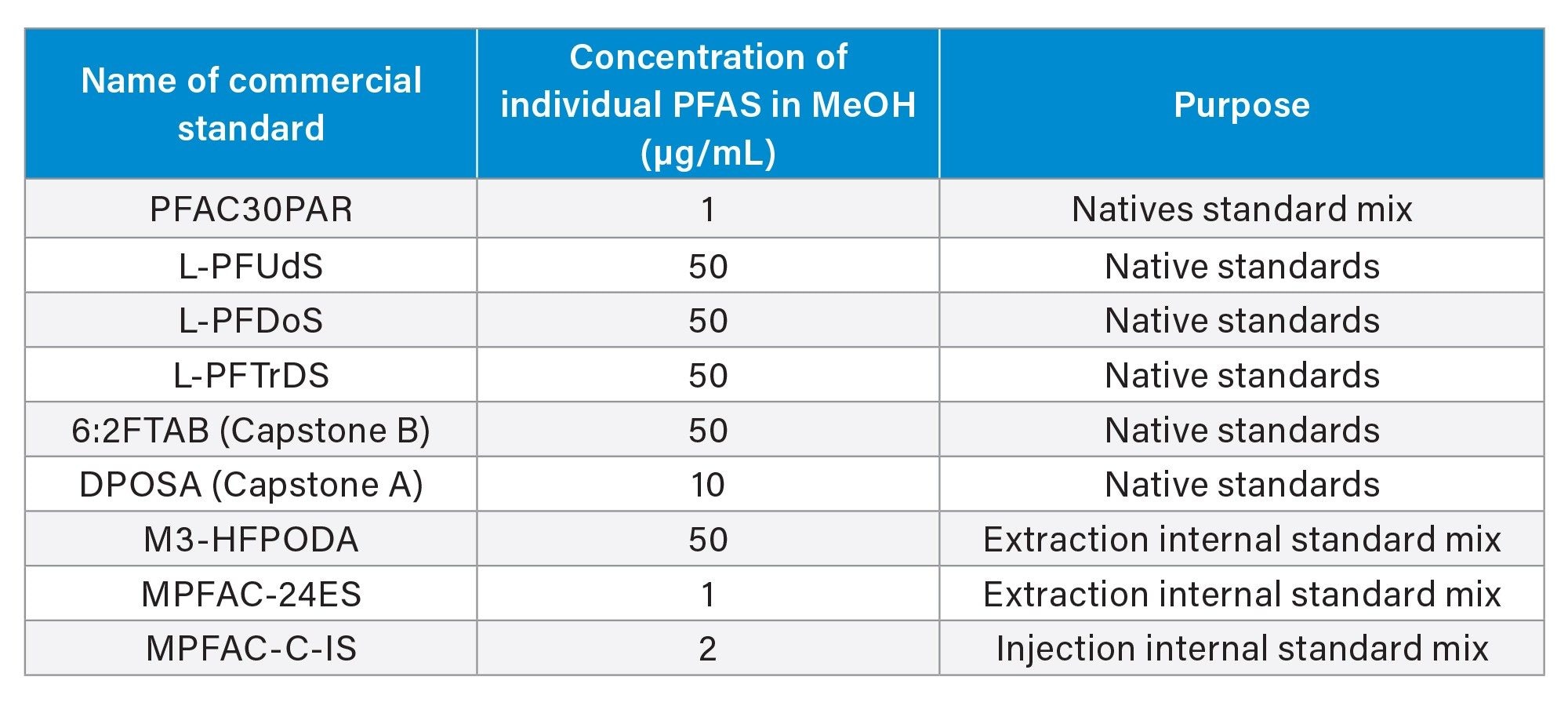
A native PFAS mix stock solution (500 ng/mL of each analyte) was prepared in methanol and was used for serial dilutions. An extraction internal standard (EIS) mix solution (1 ng/mL of each labelled analyte) was prepared in methanol and was used to spike food samples prior to extraction. A mix of EIS and injection internal standards (IIS) was prepared in water:methanol 1:1 (all proportions in this work were v:v). The EIS + IIS mix included 0.2 ng/mL of each labelled analyte; this solution served as diluent for the calibration curve. Finally, an IIS mix solution (20 ng/mL of each labelled analyte) was prepared in water:methanol 1:1 and was used to spike each LC vial after extraction and clean-up.
A solvent calibration curve in the range of 0.00125–5 ng/mL (equivalent to 0.00025–1 µg/kg in actual sample) was prepared and used for sample analysis.
Sample Preparation
Prior to any operation in the laboratory, good practices for preventing or minimising PFAS contamination from the environment and reagents was followed (details described in the White Paper 720007905).7
Test samples consisted of vegetable (tomato), fruit (apple), and baby food (an organic smooth puree made of fruit and yogurt) which were purchased at a local grocery store. The edible portions of tomato and apple were cut into thin slices, and homogenized using a kitchen blender. Samples were stored in a freezer (-20 °C) and thawed in a refrigerator (4 °C) overnight prior to extraction.
2.5 grams of sample were placed into a 15 mL centrifuge Falcon tube, spiked with 100 µL of EIS (at a concentration of 0.04 µg/kg), and vortexed for a few seconds.
The samples were extracted with 6 mL of 0.3% ammonium hydroxide methanolic solution and vortex mixing the tubes for 10 min. After centrifugation at 5000 g, the supernatant was quantitatively transferred into an empty clean tube. The extraction step was repeated, and approximately 12 mL of extract were combined and concentrated to 0.5 mL under a gentle nitrogen stream at 50 ºC. Each sample was reconstituted up to 8 mL using reagent water. The pH of each sample was checked with test strips, and corrected by adding a few drops of a 50% formic acid solution (if needed) to ensure the pH of the reconstituted extract was below 6 units.
The extract was loaded onto a prototype bilayer dual-phase SPE cartridge from Waters™ containing weak anion exchange (WAX) and graphitized carbon black (GCB). The cartridge used for clean-up of food samples has the GCB layer on top of the WAX layer. The cartridge was previously conditioned with 15 mL of 1% methanolic ammonium hydroxide, and with 5 mL of 0.3 M formic acid in water. After loading the sample, the cartridge was washed with 10 mL of reagent water and 5 mL of 1:1 0.1 M formic acid:methanol. After drying the cartridge for 10 seconds, the analytes were eluted with 5 mL of 1% methanolic ammonium hydroxide, collecting the eluate in a clean Falcon tube.
The eluate was concentrated under a gentle nitrogen stream at 50 ºC until about one drop, and then reconstituted with 0.5 mL of water:methanol 1:1. The reconstitution was performed in two steps. First 250 µL of methanol were added and the tube was vortexed. Then 250 µL of water was added and the tube was vortexed again. This approach is meant to increase the solubility of less polar PFAS.
The reconstituted cleaned-up samples (0.5 mL) were transferred into polypropylene LC vials (p/n: 186005219) with polyethylene cap (p/n: 186000305). 5 µL of IIS solution was added to each vial (0.2 ng/mL of each labelled IIS resulted in vial), which were then vortexed and placed in the autosampler for injection. The method is illustrated in Figure 1, and resulted in a concentration factor of five.
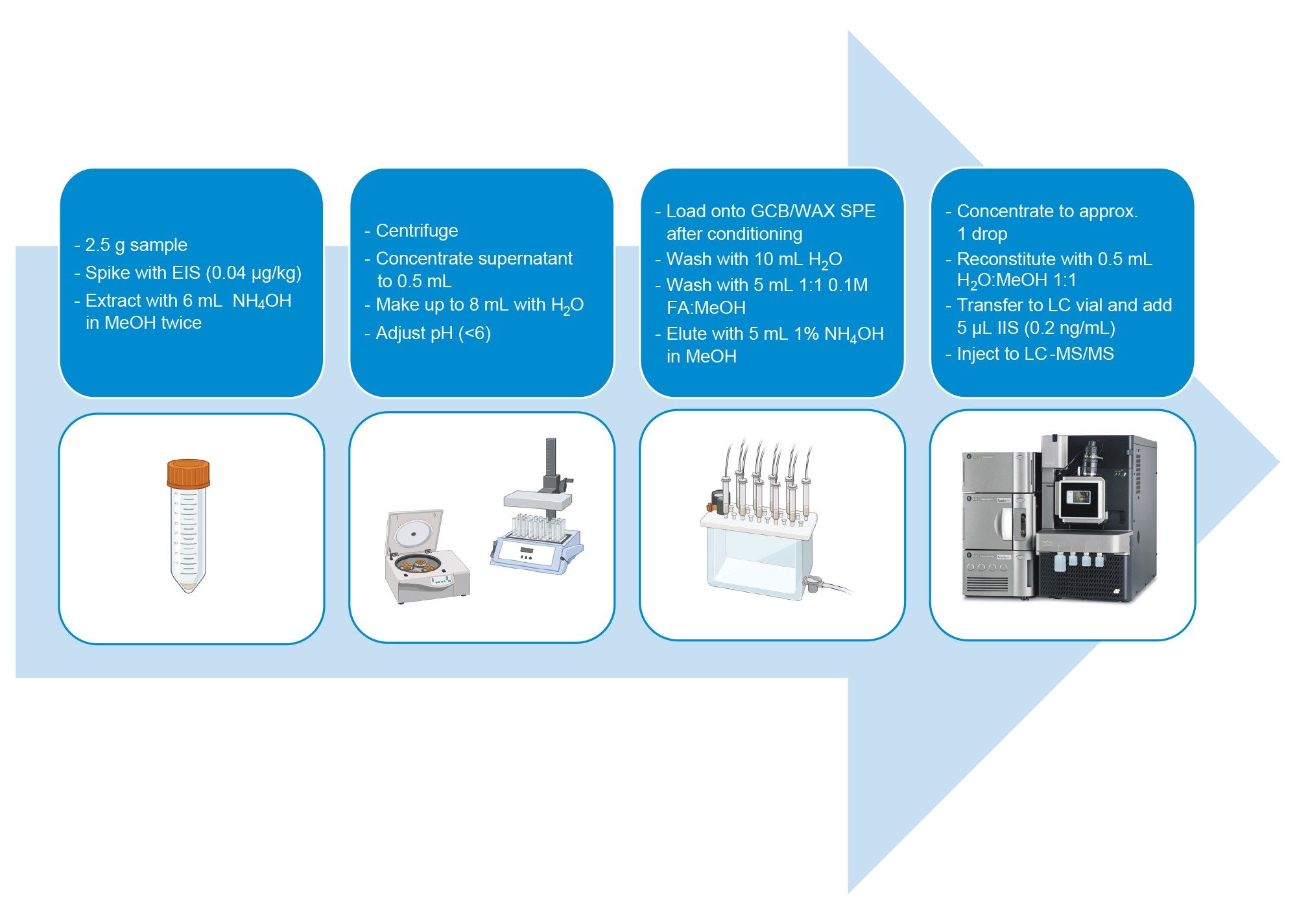 Figure 1. Scheme of the extraction and clean-up protocol for vegetable, fruit, and baby food samples.
Figure 1. Scheme of the extraction and clean-up protocol for vegetable, fruit, and baby food samples.
LC Conditions
|
LC system: |
ACQUITY™ PREMIER UPLC with PFAS Analysis Kit |
|
Vials: |
Polypropylene autosampler vial (p/n: 186005219) with polyethylene cap (p/n: 186000305) |
|
Analytical column: |
ACQUITY Premier UPLC BEH™ C18, 2.1 x 50 mm, 1.7 µm (p/n: 186009452) |
|
Isolator column: |
Atlantis™ Premier BEH C18 AX 2.1 x 50 mm, 5.0 µm (p/n: 186009407) |
|
Column temperature: |
35 °C |
|
Sample temperature: |
10 °C |
|
Injection volume: |
5 µL |
|
Flow rate: |
0.3 mL/min |
|
Mobile phase A: |
2 mM ammonium acetate in water |
|
Mobile phase B: |
2 mM ammonium acetate in methanol/acetonitrile 1/1 (v/v) |
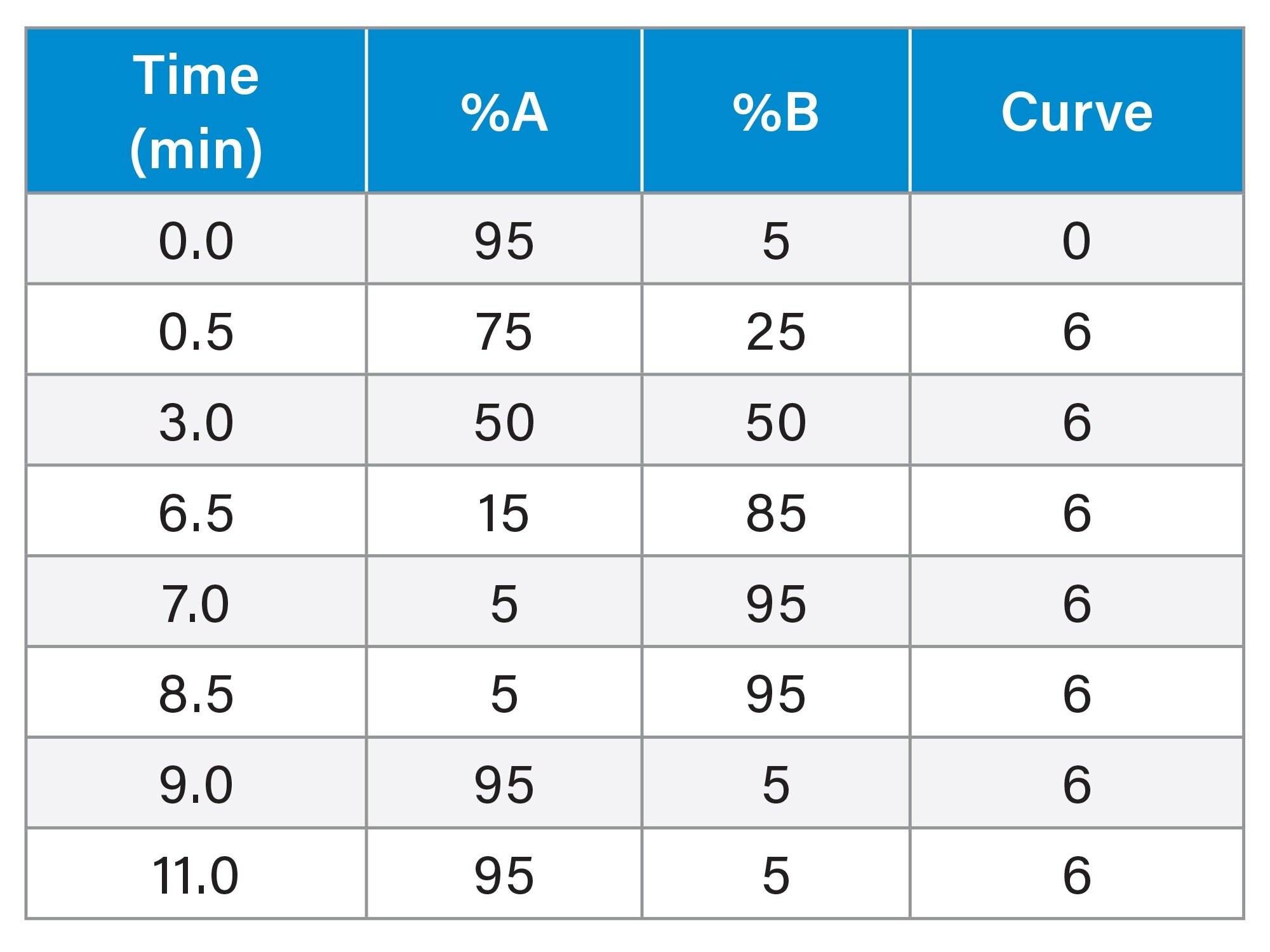
Gradient Table
MS Conditions
|
MS system: |
Xevo TQ Absolute |
|
Ionization mode: |
Electrospray negative |
|
Source temperature: |
100 °C |
|
Capillary voltage: |
0.5 kV |
|
Desolvation temperature: |
350 °C |
|
Desolvation flow rate: |
900 L/hr |
|
Cone flow rate: |
150 L/hr |
|
MRM method: |
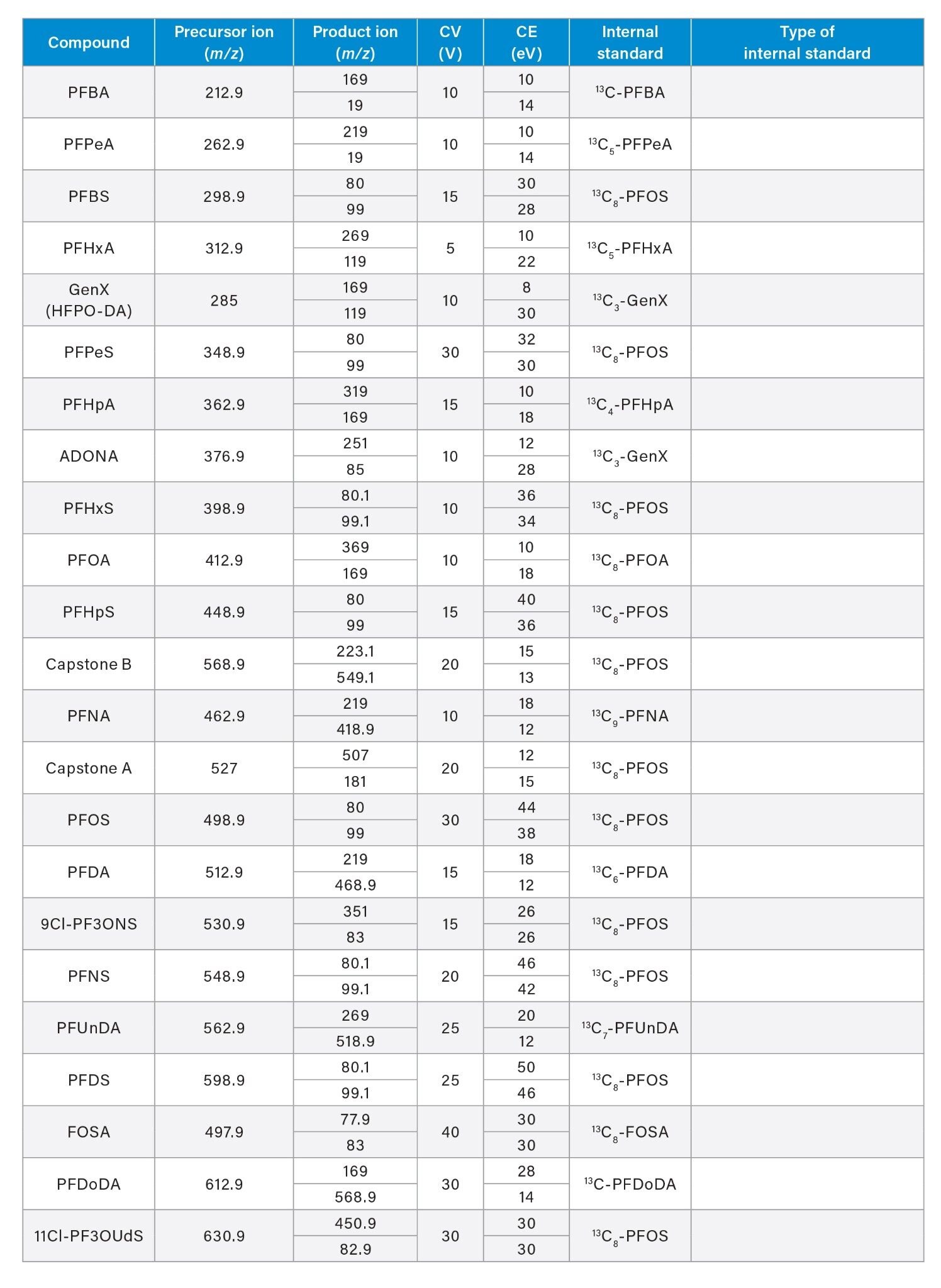
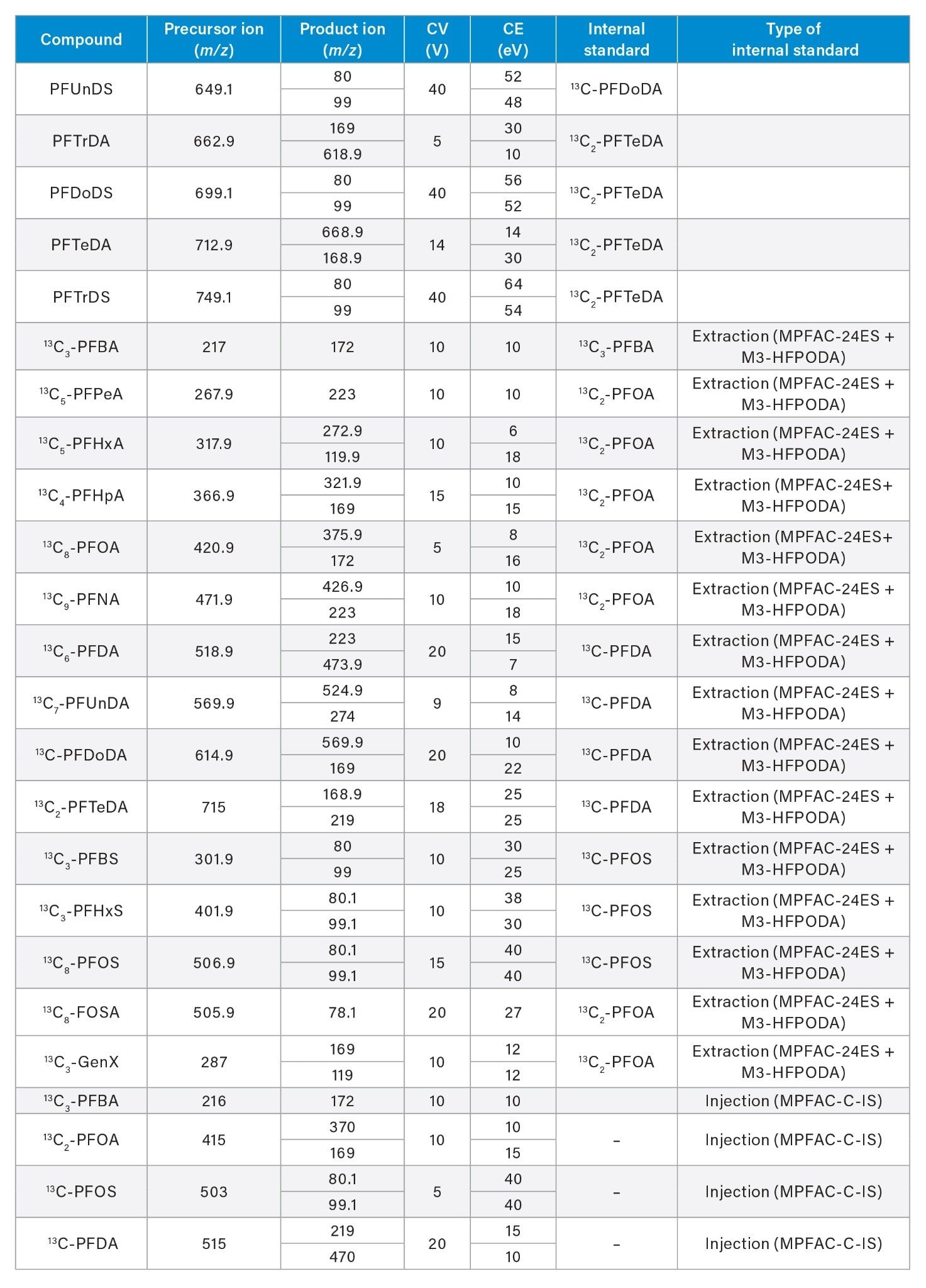
See Appendix for Full MRM Method details |
Data Management
|
Software: |
waters_connect™ for quantitation |
Method Performance Assessment
The performance of the method was evaluated using the criteria set in the EURL POPs PFAS guidance document and AOAC SMPR.4,6 For the intra-lab method performance, trueness was assessed by spiking samples of tomato, apple, and baby food with a mixture of 28 native PFAS at three concentration levels, in triplicate. Solvent blanks, procedural blanks, matrix blanks, and fortified samples were then extracted and analysed as described in the previous section. Repeatability of the method was assessed by relative standard deviation under repeatability conditions intra-day (RSDr). Apparent recoveries and RSDr were calculated for each level:
Level 1: corresponding to the method-lower limit of quantification (m-LLOQ)
Level 2: corresponding to 10x m-LLOQ
Level 3: corresponding to the method-upper limit of quantification (m-ULOQ)
The analytical sequence of each batch was composed of two different sets for solvent calibrants (≥6 points per each curve, excluding blank) bracketing incurred, and fortified samples.
Results and Discussion
The method includes 28 PFAS that are listed in the Commission Recommendation (EU) 2022/1431, including mandatory, recommended and PFAS “for consideration” (see Table 1 for the list of PFAS).
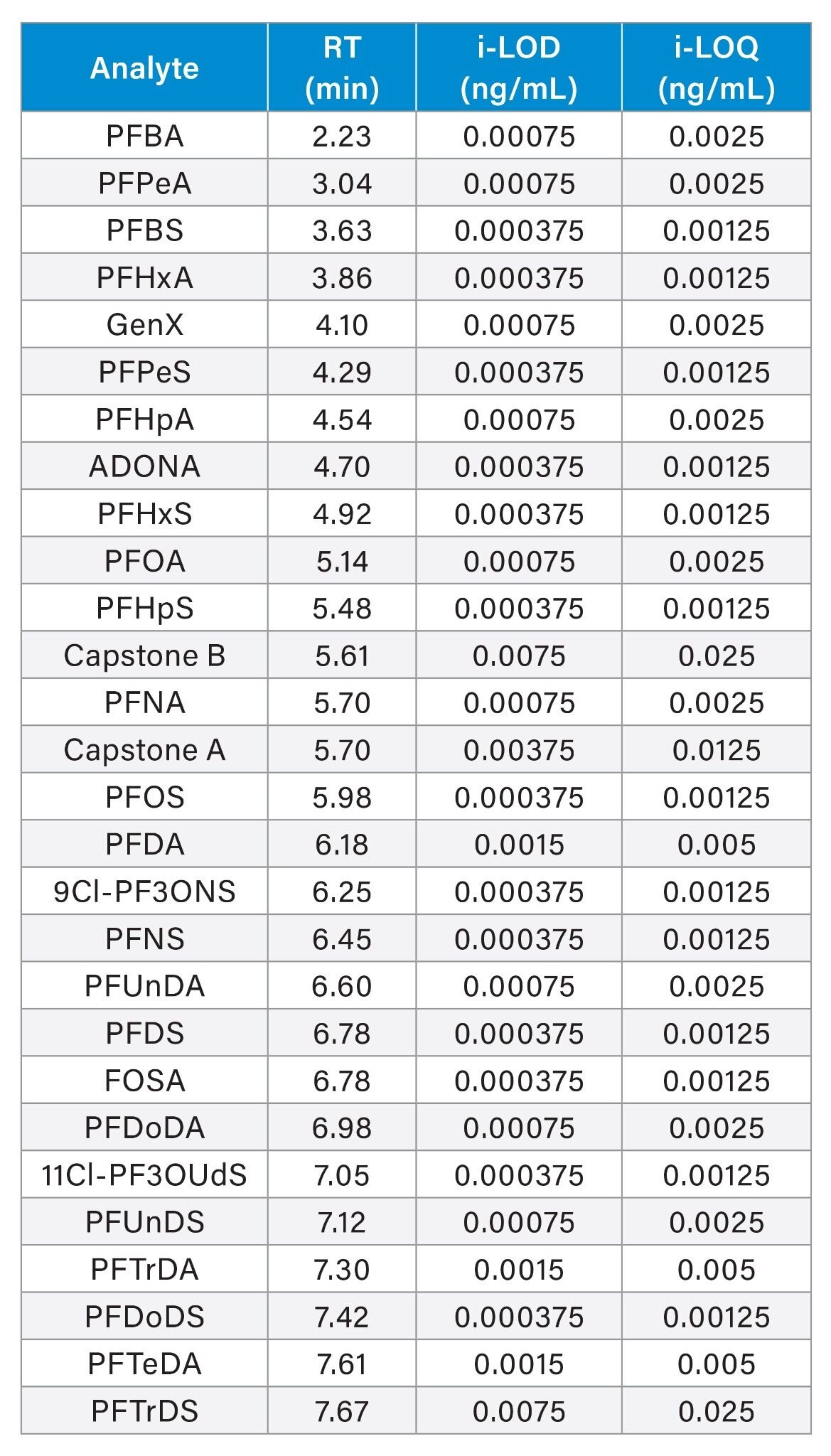 Table 1. Retention time (RT), instrument-limit of detection (i-LOD) and instrument-limit of quantification (i-LOQ) of 28 PFAS compounds listed in the Commission Recommendation (EU) 2022/1431 using the Xevo-TQ Absolute Mass Spectrometer.
Table 1. Retention time (RT), instrument-limit of detection (i-LOD) and instrument-limit of quantification (i-LOQ) of 28 PFAS compounds listed in the Commission Recommendation (EU) 2022/1431 using the Xevo-TQ Absolute Mass Spectrometer.The EIS were spiked prior to sample extraction and used to correct the native compounds for extraction recovery and matrix effects. The IIS were added to the sample after clean-up and used to correct the extraction standards for reconstitution variations, matrix effects, and potential injection variations. With the presence of EIS and IIS, matrix-matching was not necessary for routine sample analysis. This approach has been discussed in a previous study.8
Chromatography
Baseline separation of linear and branched-PFAS was achieved on a 50 mm BEH C18 column after scaling down a previously developed LC method from 22 to 11 minutes of run time. A combination of methanol and acetonitrile in the organic mobile phase was used to obtain baseline separation of PFAS from potential interferences commonly found in certain types of foods (e.g., separating cholic acids from PFOS). Figure 2 shows the chromatographic trace of PFHxS and PFOS, where it can be noted that linear and branched PFAS can be quantified either individually or as a sum given to the excellent chromatographic separation. More information regarding LC optimization can be found in our previous work App Note 720008108.9
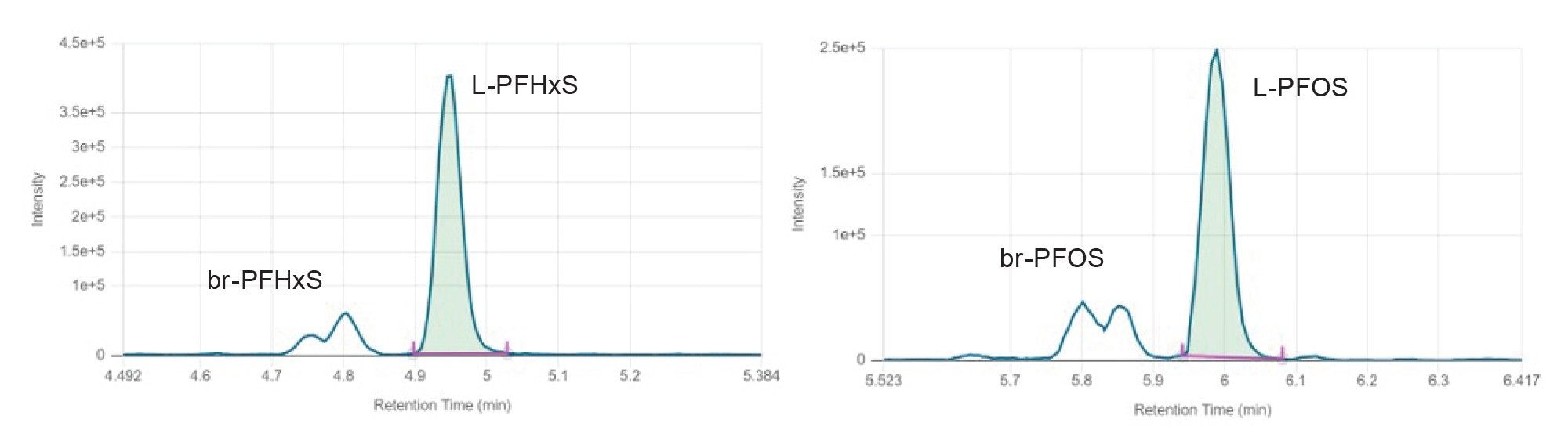 Figure 2. Chromatographic traces of a tomato sample spiked with 0.01 µg/kg of PFHxS (on the left) and PFOS (on the right). L=linear, br=branched.
Figure 2. Chromatographic traces of a tomato sample spiked with 0.01 µg/kg of PFHxS (on the left) and PFOS (on the right). L=linear, br=branched.
The ratio of the chromatographic retention time (RT) of the analyte to that of the IS (i.e. relative RT of the analyte) corresponded to that of the calibration standard with a deviation ≤1%.
The first eluting compound (PFBA, RT=2.23 min) presents a RT >6 times the column void time (t0=0.37), thus enough retention was obtained for all PFAS included in this study.
Linearity and Limits of Quantification
The response of solvent standards, which bracketed the incurred and fortified samples, were plotted to construct the calibration curve using 1/x weighting factor. The linear range of the method differed across different analytes and matrices. Coefficients of determination of the calibration curves (R2) were >0.99, and residuals were within ±20% in the majority of the cases. Ion ratios from sample extracts were within ±30% (relative) of average of calibration standards from same sequence.
The instrument limit of detection (i-LOD)10 was calculated on solvent standards (signal-to-noise ratio ≥3 at the LOD level) and were as low as 0.0004 ng/mL for certain individual PFAS (equivalent to 2 fg on column).
The method lower and upper limits of quantification (m-LLOQ and m-ULOQ) were adopted as the lowest and highest level of the calibration range, respectively, provided that m-LLOQ presented a signal-to-noise ratio ≥10 (noise processing=peak-to-peak). m-LLOQ were as low as 0.0005 µg/kg in the tested samples. i-LOD, i-LOQ and retention times are reported in Table 1, while m-LLOQ, method linear range and linearity for each food commodity can be found in Tables 2, 3, and 4.
Notably, for the four mandatory PFAS (PFOS, PFOA, PFNA, and PFHxS) the method allows to accurately quantify as low as 0.0005 µg/kg in all food commodities tested. Figure 3 presents the calibration curves and residuals plot, whilst Figure 4 shows an example chromatogram at the m-LLOQ level.
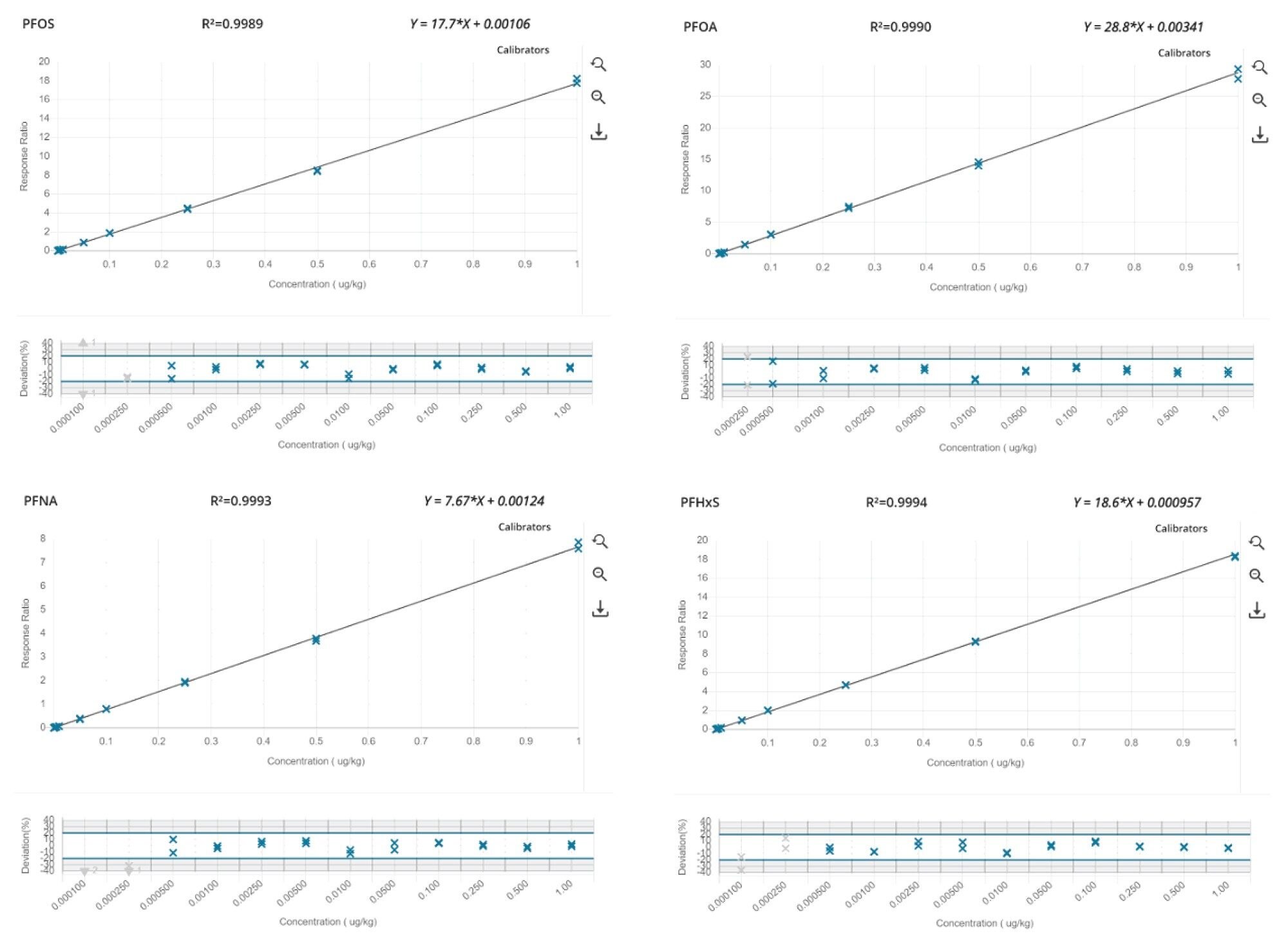 Figure 3. Calibration curves and residuals plots of the EU mandatory PFAS (PFOS, PFOA, PFNA, and PFHxS).
Figure 3. Calibration curves and residuals plots of the EU mandatory PFAS (PFOS, PFOA, PFNA, and PFHxS).
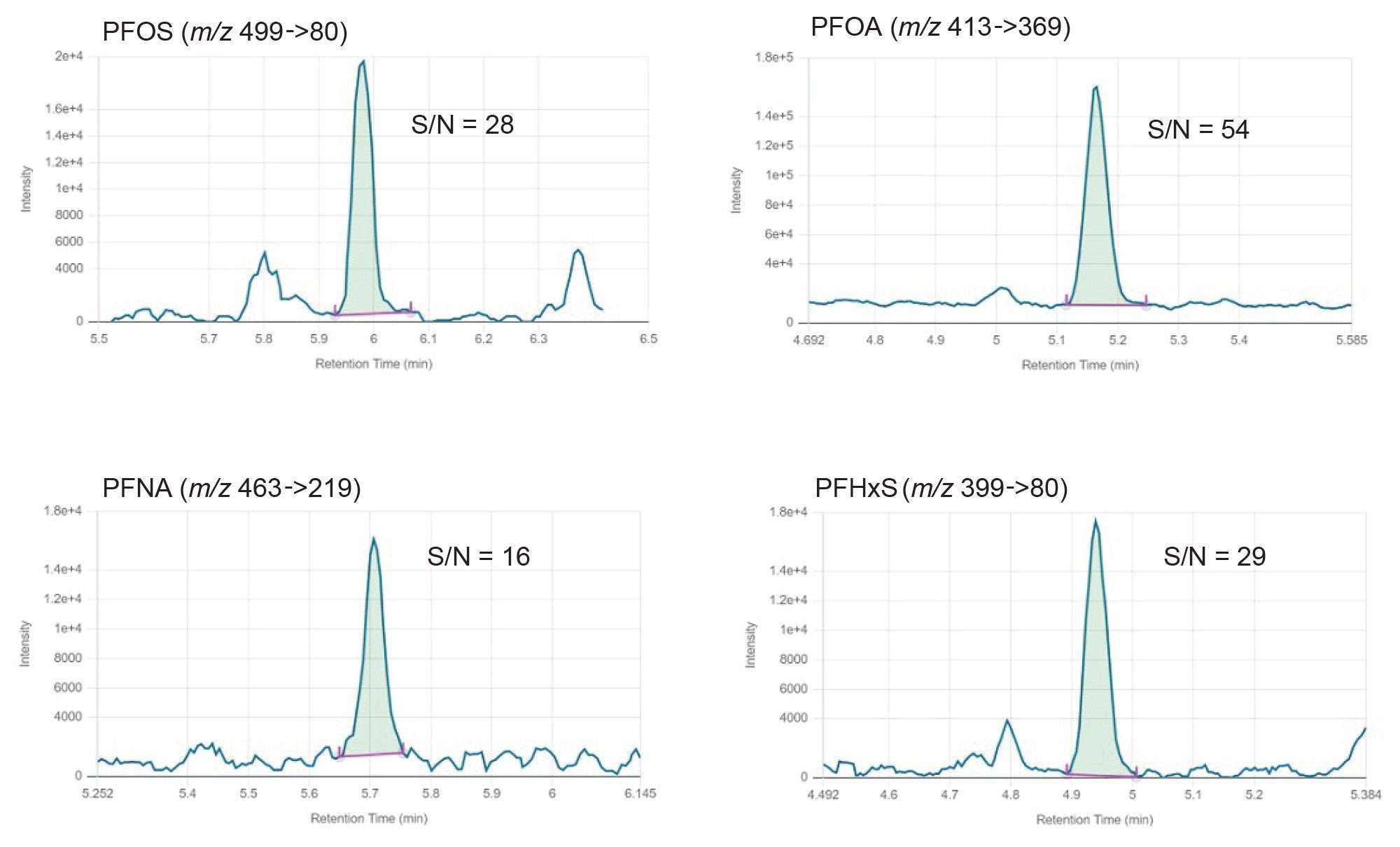 Figure 4. Chromatographic traces of 0.0005 µg/kg of EU mandatory PFAS in an apple sample. S/N: signal-to-noise ratio (noise processing=peak-to-peak).
Figure 4. Chromatographic traces of 0.0005 µg/kg of EU mandatory PFAS in an apple sample. S/N: signal-to-noise ratio (noise processing=peak-to-peak).
Trueness and Repeatability
Trueness, determined as apparent recovery, was assessed by recovery experiments which involved spiking a blank sample at three concentration levels. When considering the entire panel of native PFAS together, mean percentage recovery across all fortification levels was 101±14% for tomato (min=65%, max=131%), 98±13% for apple (min=65%, max=131%), and 99±13% for baby food (min=66%, max=127%). For the four mandatory compounds (PFOS, PFOA, PFNA, and PFHxS) apparent recoveries were between 84 and 116%. For the remaining PFAS recommended and considered in Commission Recommendation (EU) 2022/1431 for monitoring purposes, apparent recoveries were within 65 and 131%. Both sets of results meet the acceptance criteria for trueness described in the CIR, EURL POPS guidelines, and AOAC SMPR. Repeatability (RSDr) ranged between 0.4 and 10% across all matrices and at all fortification levels, within the limits described for RSDr in the AOAC SMPR*. The only exceptions were PFDS, 9Cl-PF3ONS, and 11Cl-PF3OUdS, whose recoveries in apple and baby food were within the criteria, but in tomato recoveries were 53, 52, and 58%, respectively. Nevertheless, since RSDr were well below 10%, for these three compounds, it could be possible to apply a recovery correction factor, as precision is not compromised. Recoveries and RSDr are reported in Tables 2, 3, and 4, and plotted in Figures 5, 6, and 7.
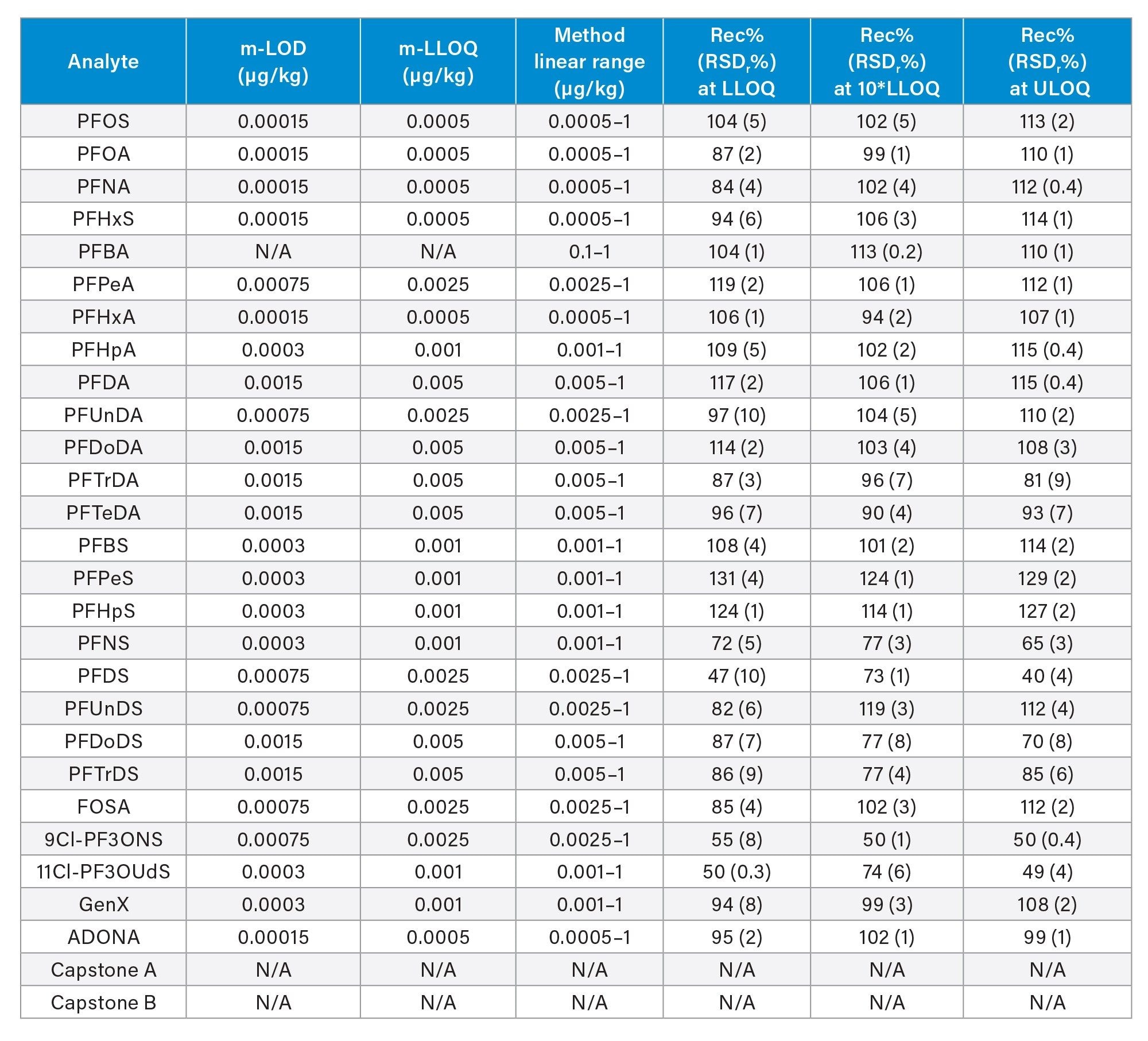 Table 2. Method performance parameters in tomato: method limit of detection (m-LOD) and method lower limit of quantification (m-LLOQ), linear range, percentage recovery at three levels, and respective percentage relative standard deviation under repeatability conditions (RSDr%, n=3). ULOQ=method upper limit of quantification (equivalent to the highest level of the calibration curve).
Table 2. Method performance parameters in tomato: method limit of detection (m-LOD) and method lower limit of quantification (m-LLOQ), linear range, percentage recovery at three levels, and respective percentage relative standard deviation under repeatability conditions (RSDr%, n=3). ULOQ=method upper limit of quantification (equivalent to the highest level of the calibration curve).
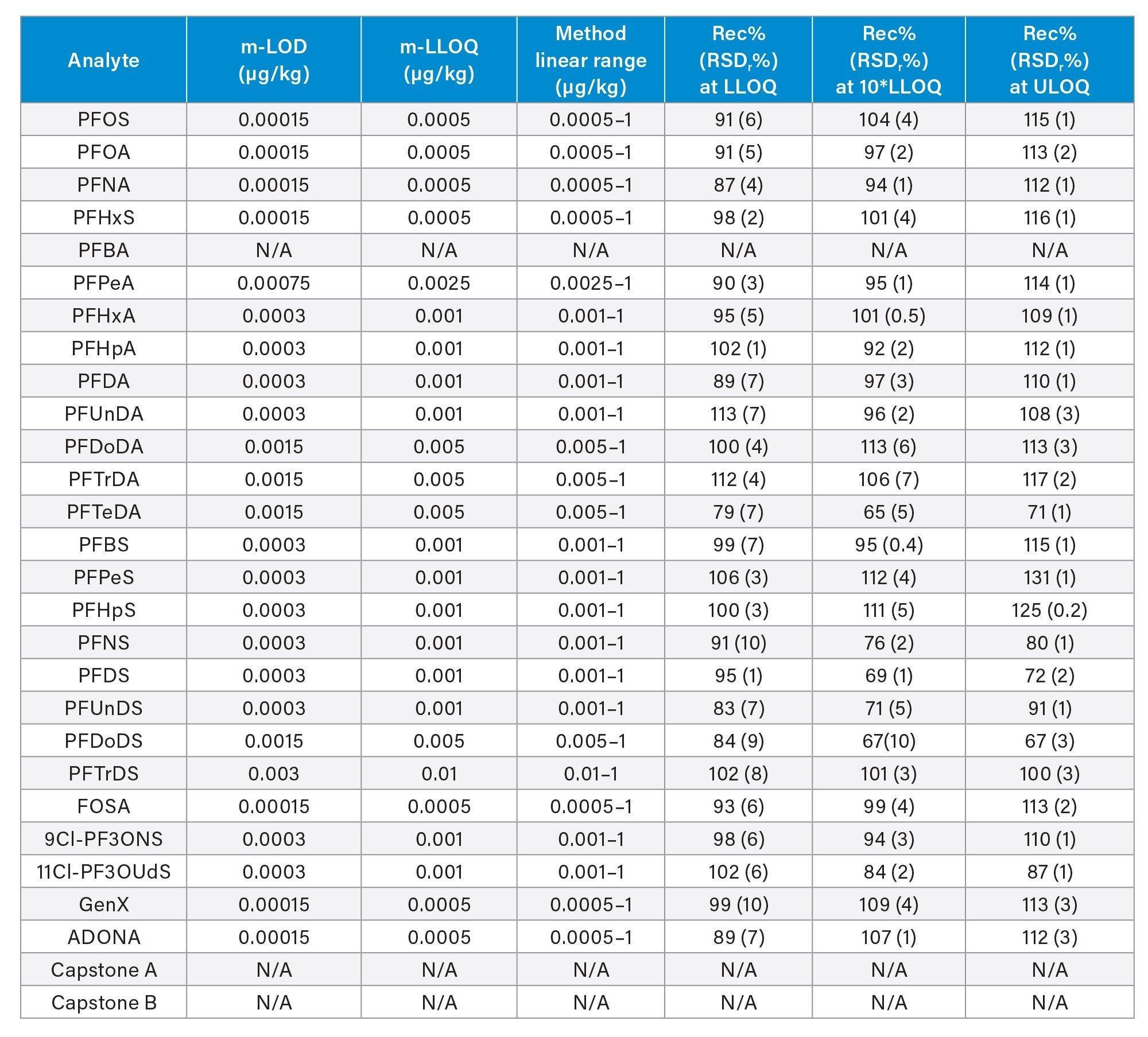 Table 3. Method performance parameters in apple: method limit of detection (m-LOD) and method lower limit of quantification (m-LLOQ), linear range, percentage recovery at three levels and respective percentage relative standard deviation under repeatability conditions (RSDr%, n=3). ULOQ=method upper limit of quantification (equivalent to the highest level of the calibration curve).
Table 3. Method performance parameters in apple: method limit of detection (m-LOD) and method lower limit of quantification (m-LLOQ), linear range, percentage recovery at three levels and respective percentage relative standard deviation under repeatability conditions (RSDr%, n=3). ULOQ=method upper limit of quantification (equivalent to the highest level of the calibration curve).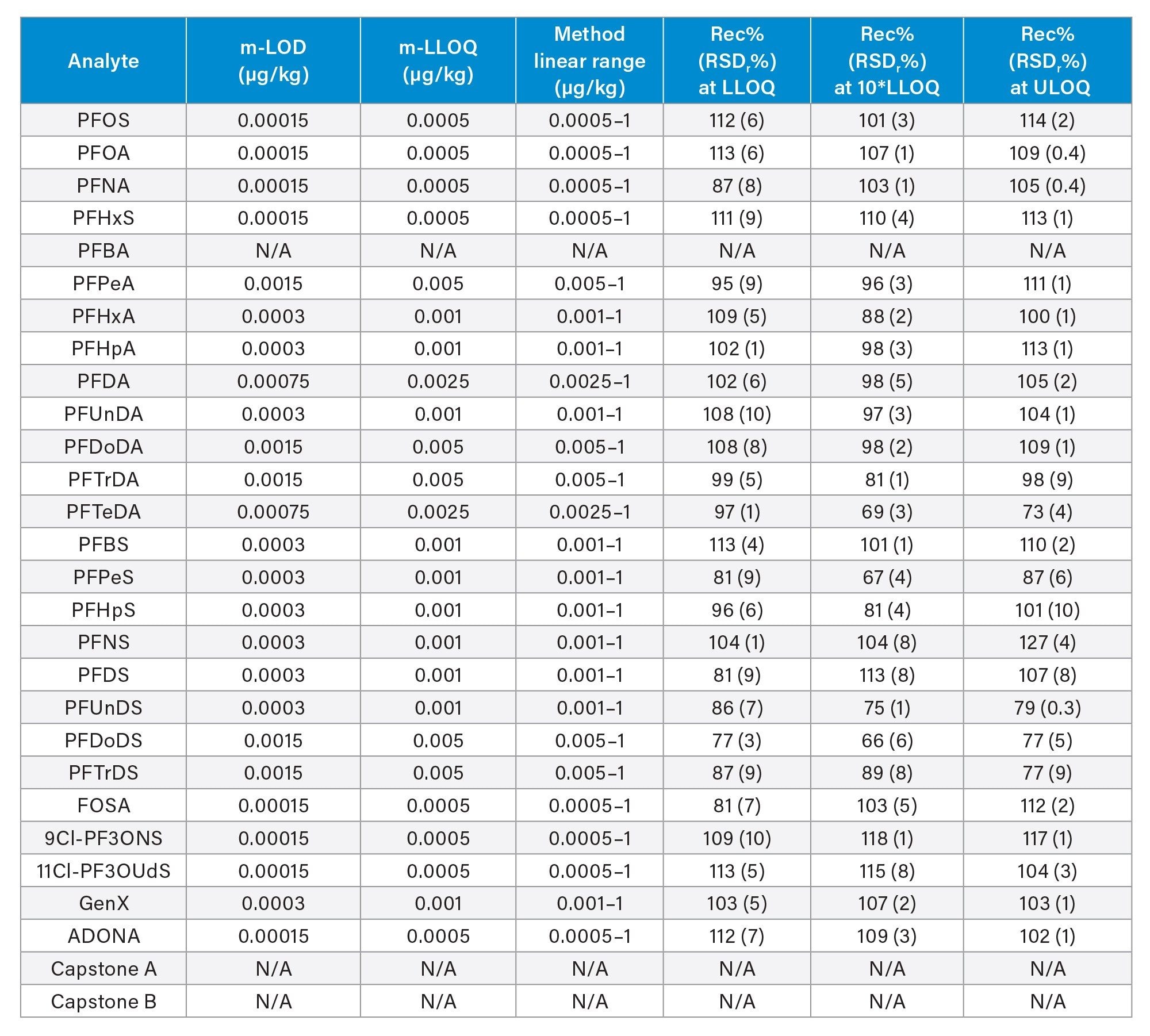 Table 4. Method performance parameters in baby food: method limit of detection (m-LOD) and method lower limit of quantification (m-LLOQ), linear range, percentage recovery at three levels and respective percentage relative standard deviation under repeatability conditions (RSDr%, n=3). ULOQ=method upper limit of quantification (equivalent to the highest level of the calibration curve).
Table 4. Method performance parameters in baby food: method limit of detection (m-LOD) and method lower limit of quantification (m-LLOQ), linear range, percentage recovery at three levels and respective percentage relative standard deviation under repeatability conditions (RSDr%, n=3). ULOQ=method upper limit of quantification (equivalent to the highest level of the calibration curve).(*) No assessment of intermediate precision (RSDR) was made during this study.
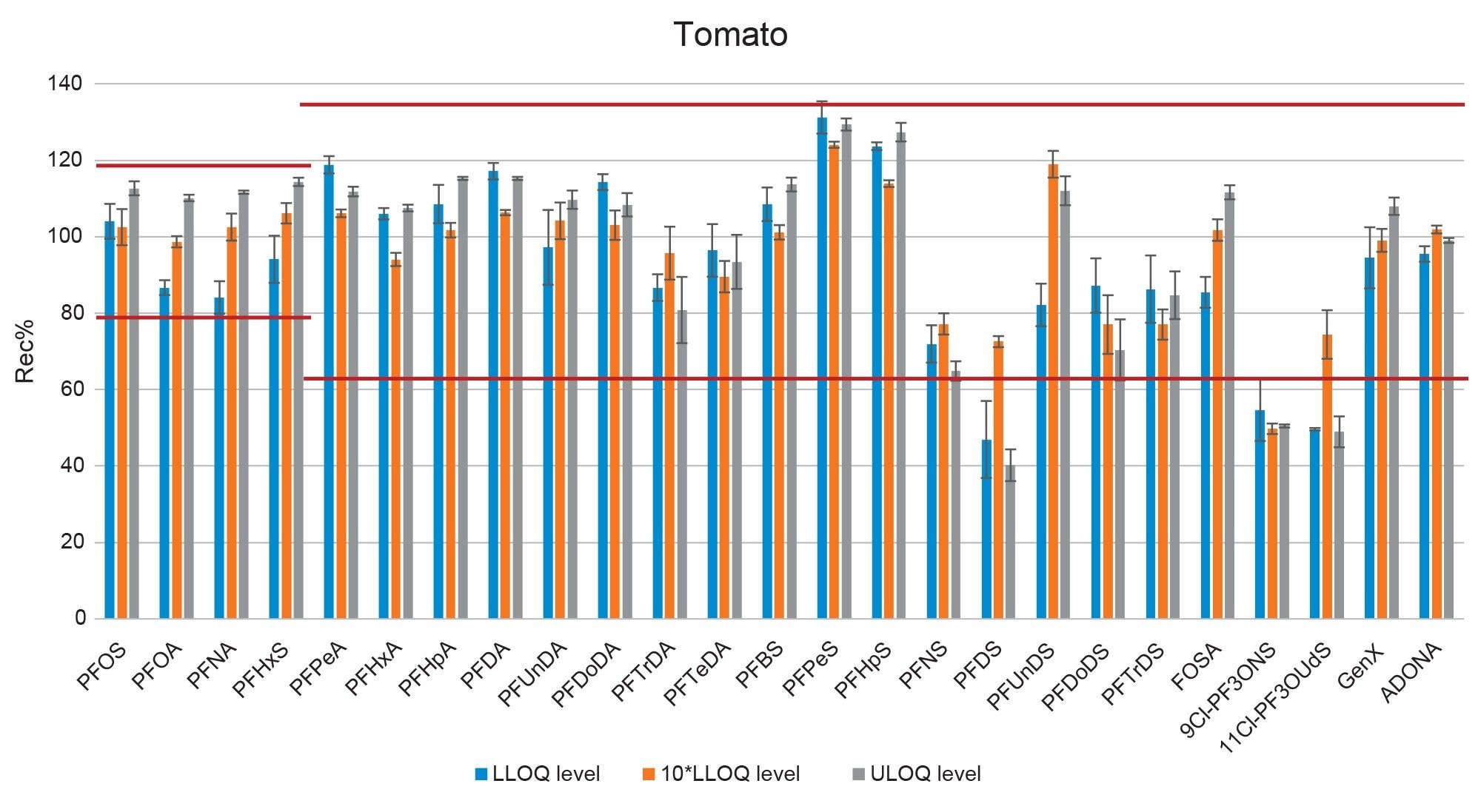 Figure 5. Bar-plot representing the recovery of PFAS in tomato at three fortification levels. Red lines represent the thresholds set by the EURL POPs guidelines.
Figure 5. Bar-plot representing the recovery of PFAS in tomato at three fortification levels. Red lines represent the thresholds set by the EURL POPs guidelines.
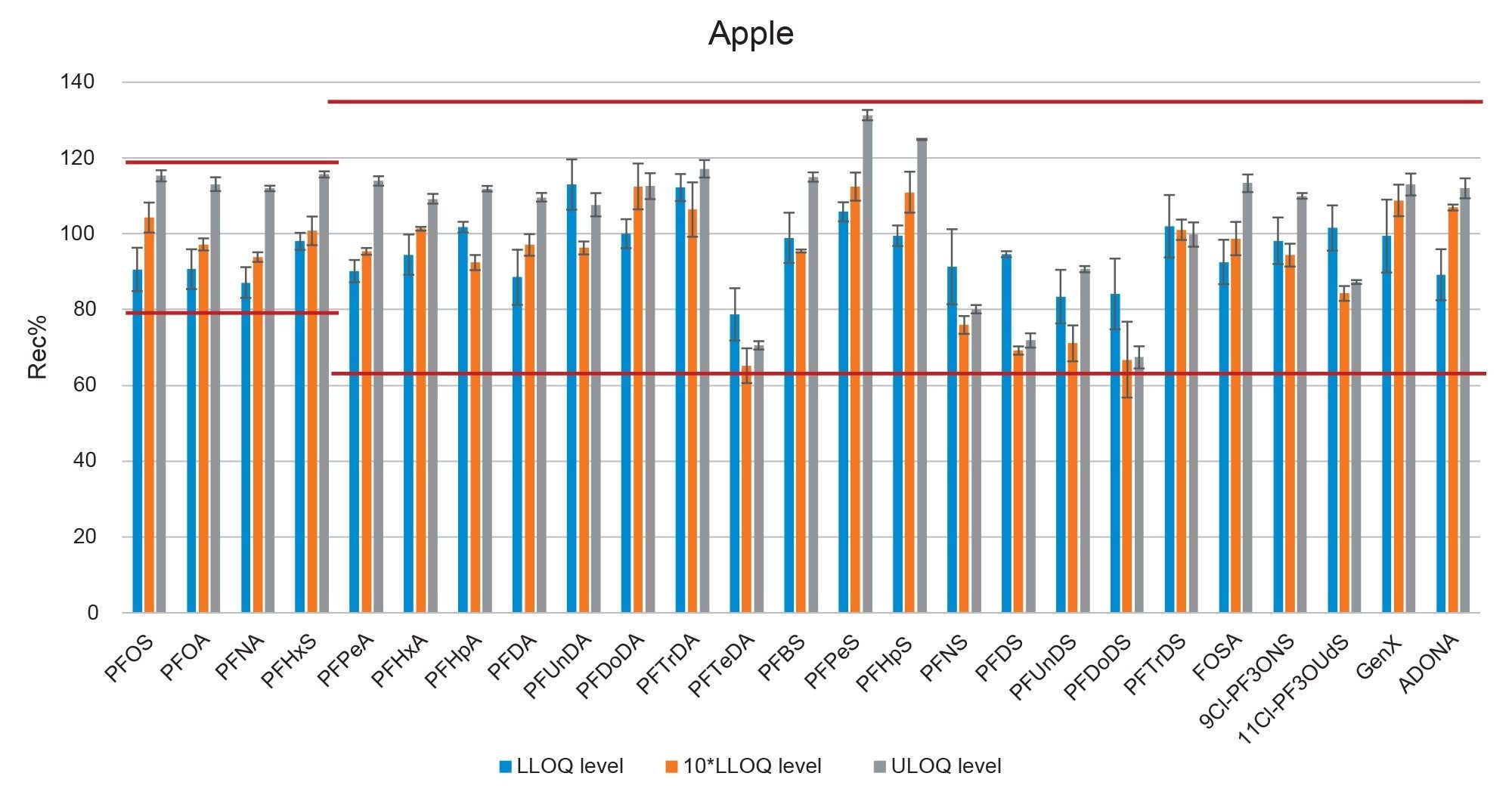 Figure 6. Bar-plot representing the recovery of PFAS in apple at three fortification levels. Red lines represent the thresholds set by the EURL POPs guidelines.
Figure 6. Bar-plot representing the recovery of PFAS in apple at three fortification levels. Red lines represent the thresholds set by the EURL POPs guidelines.
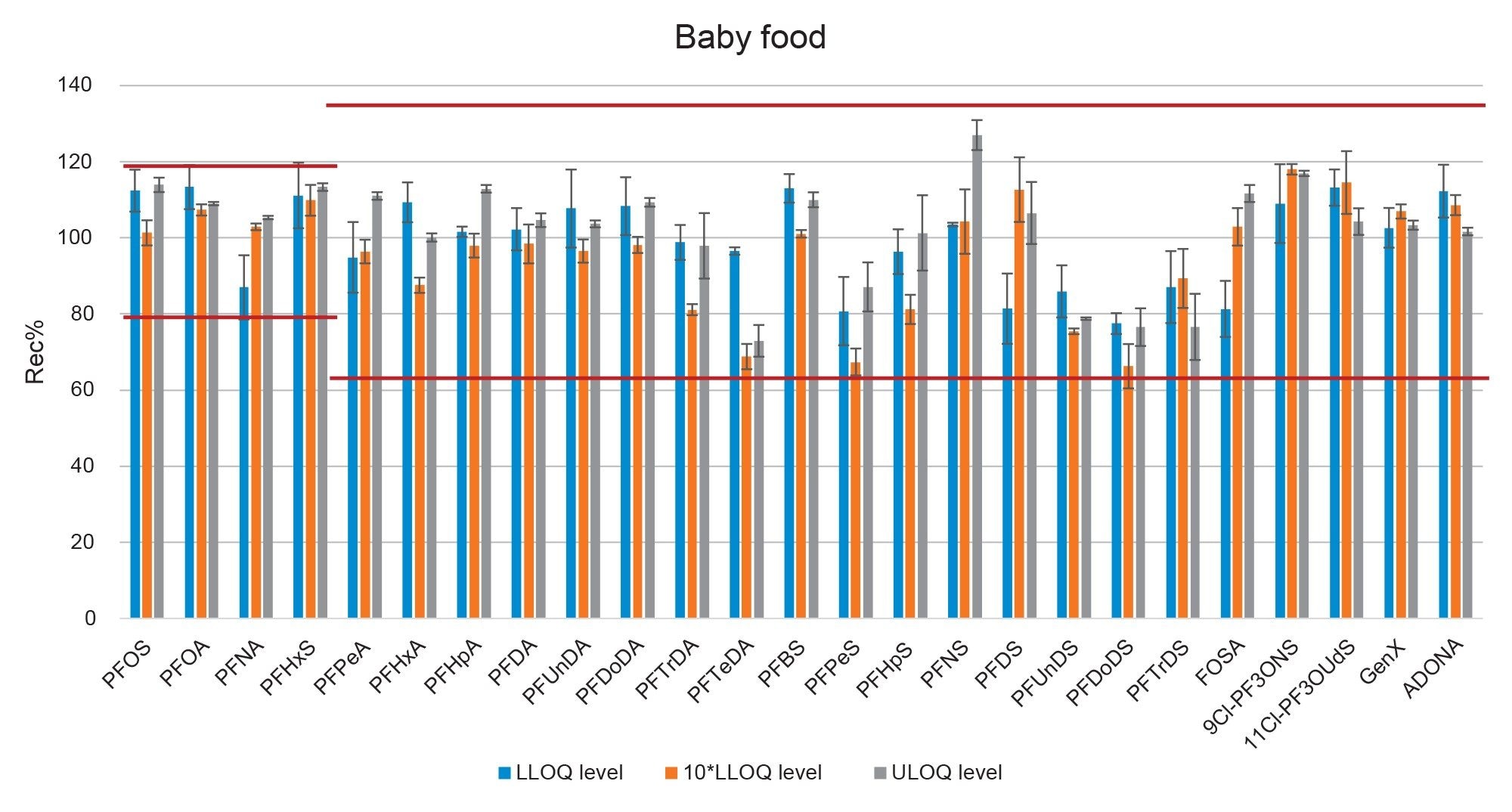 Figure 7. Bar-plot representing the recovery of PFAS in baby food at three fortification levels. Red lines represent the thresholds set by the EURL POPs guidelines.
Figure 7. Bar-plot representing the recovery of PFAS in baby food at three fortification levels. Red lines represent the thresholds set by the EURL POPs guidelines.
During this study, a remarkably high contamination of PFBA was encountered not only in matrix samples, but also in solvents and reagents such as formic acid and methanol. PFBA was found in procedural blanks and in both tomato and apple at 0.2 µg/kg. More worryingly, PFBA was also found in baby food at an estimated concentration of 17.5 µg/kg (extrapolated from the calibration curve). PFBA contamination varied from batch to batch, but was always present in all types and brands of methanol that were tested, making it impossible to obtain accurate recovery data for this compound in the present work.
Many PFAS compounds were also found in matrix blanks at levels close to the m-LLOQ. In such cases, blank subtraction was performed in the calculation of recovery.
Capstone A and B were investigated within the method development phase, and appeared to yield to very low process recoveries (below 10%). To better understand the causes of this, an experiment was conducted in which a solvent standard at 0.5 ng/mL was loaded onto the GCB/WAX cartridge after conditioning, and each fraction of the SPE process was collected and analysed after dilution with water or methanol to obtain a final composition of 1:1 water:methanol in the vial. As it can be seen in Figure 8, about 31 and 37% of the analyte was lost in the second washing step for Capstone A and B, respectively. Some researchers11 also found that these zwitterionic compounds were not efficiently retained on a GCB/WAX SPE cartridge, and suggested to separate capstones from the other PFAS, and to analyse capstones via an extraction-dilution-injection approach. Another factor contributing to these low recoveries could be the impact of potential ion suppression phenomena in ESI affecting these compounds. Furthermore, as no labelled internal standards were commercially available for capstone A and capstone B, and due to their peculiar chemical structure, to achieve a more accurate quantification a standard addition approach could be carried out.
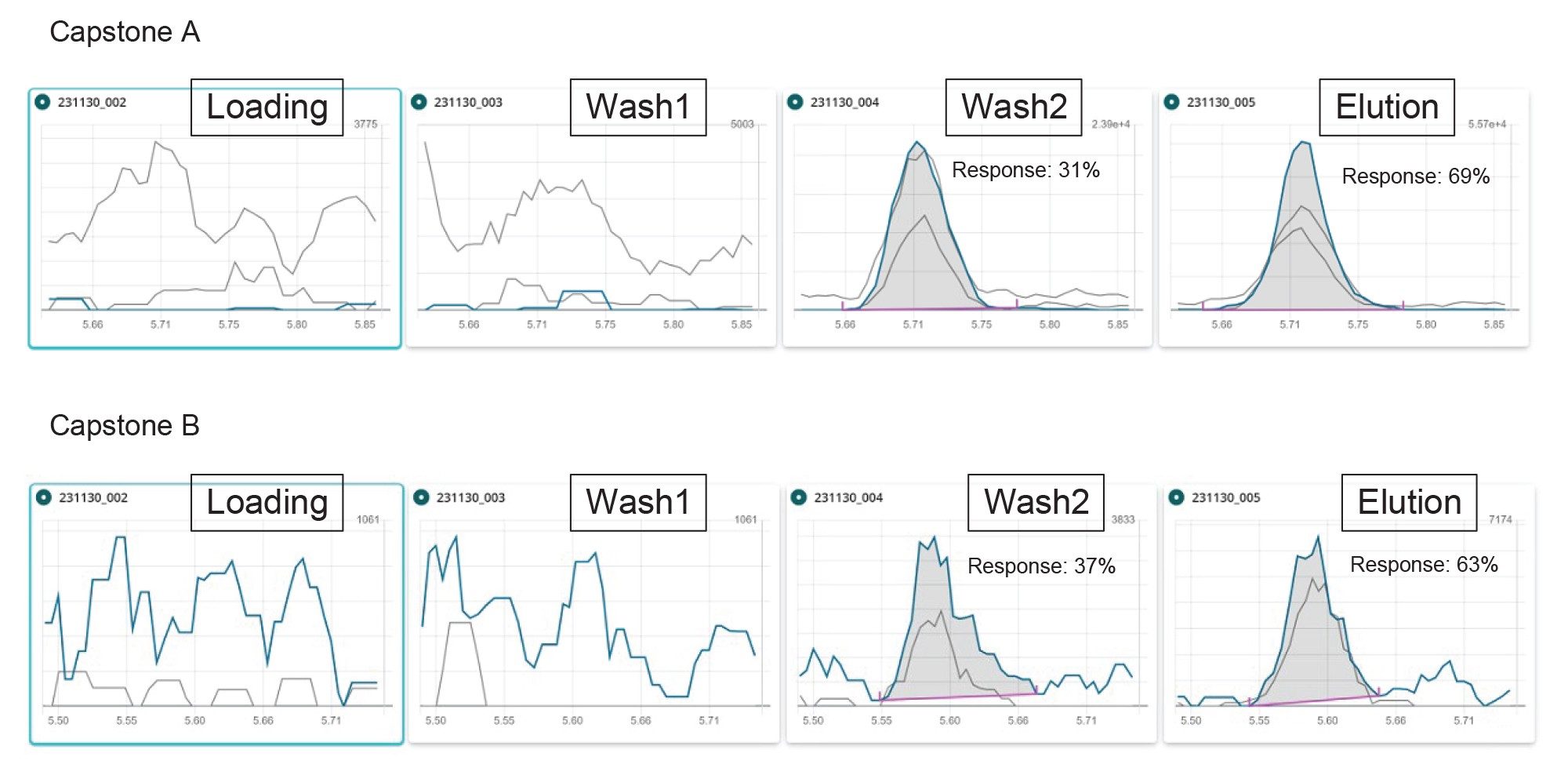 Figure 8. Chromatograms of capstone A (top) and capstone B (bottom), for each fraction of the SPE protocol. Wash1=wash step with water, wash2=wash step with 1:1 0.1 M formic acid:methanol. Blue trace: quantifier transition, grey traces: qualifier transitions.
Figure 8. Chromatograms of capstone A (top) and capstone B (bottom), for each fraction of the SPE protocol. Wash1=wash step with water, wash2=wash step with 1:1 0.1 M formic acid:methanol. Blue trace: quantifier transition, grey traces: qualifier transitions.
Notably, the recoveries for FOSA were in the range 85–112%, indicating that the sample preparation and clean-up approach herein presented can be suitable for the determination of neutral PFAS, in addition to the ionic PFAS. A similar experiment to the one described for capstones has been carried out for FOSA, showing almost full recovery of this compound in the eluate fraction (see Figure 9).
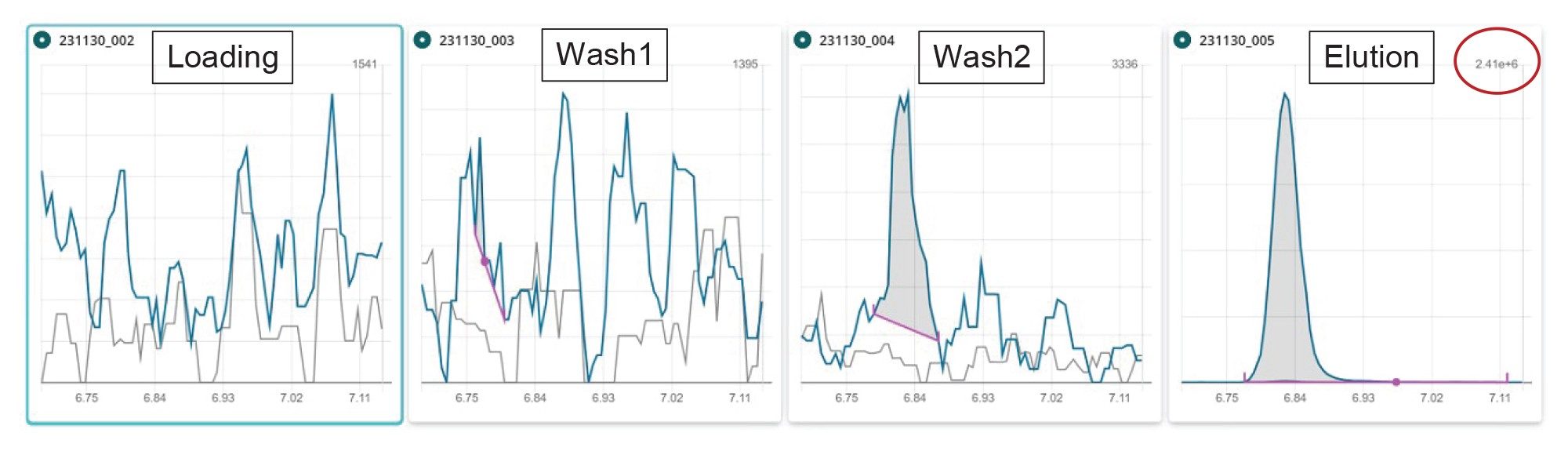 Figure 9. Chromatograms of FOSA for each fraction of the SPE protocol. Wash1=wash step with water, wash2=wash step with 1:1 0.1 M formic acid:methanol. Blue trace: quantifier transition, grey traces: qualifier transitions.
Figure 9. Chromatograms of FOSA for each fraction of the SPE protocol. Wash1=wash step with water, wash2=wash step with 1:1 0.1 M formic acid:methanol. Blue trace: quantifier transition, grey traces: qualifier transitions.
Conclusion
This study presents an optimized LC-MS/MS method for the comprehensive analysis of PFAS in vegetable, fruit, and baby food. The method demonstrated an exceptionally low limit of quantification (down to 0.0005 µg/kg for some compounds) accurately detecting and quantifying the PFAS compounds listed in the Commission Recommendation (EU) 2022/1431.
The increased cleanup efficacy of the new GCB/WAX SPE cartridge, in combination with the enhanced sensitivity of the Xevo TQ Absolute MS System, produced excellent recoveries, between 87 and 116% for the mandatory PFAS, and between 65 and 131% for the majority of the recommended and considered compounds, with RSDr ≤10%.
The method also demonstrated excellent recoveries for FOSA, a neutral PFAS which was usually very challenging to retain onto weak anionic exchange cartridges.
Limitations were observed in the analysis of capstone A and B, due to their complex zwitterionic structure, and for PFBA, due to contamination in solvents and reagents. Therefore, more work should be done to investigate on best approaches to reduce PFBA contamination, as well as to obtain higher recoveries for capstones.
The second part of the present study will concern a workflow for the determination of PFAS in animal products.
References
- Commission Regulation (EU) 2022/2388, amending Regulation (EC) No 1881/2006 as regards maximum levels of perfluoroalkyl substances in certain foodstuffs, L 316/38, 8.12.2022.
- Commission Recommendation (EU) 2022/1431, on the monitoring of perfluoroalkyl substances in food. L 221/105, 26.8.2022.
- Commission Implementing Regulation (EU) 2022/1428, laying down methods of sampling and analysis for the control of perfluoroalkyl substances in certain foodstuffs. L 221/66, 26.08.2022.
- EURL for halogenated POPs in feed and food (2022): Guidance Document on Analytical Parameters for the Determination of Per- and Polyfluoroalkyl Substances (PFAS) in Food and Feed, version 1.2 of 11 May 2022. Available online under https://eurl-pops.eu/core-working-groups#_pfas.
- EURL for halogenated POPs in feed and food (2022): Guidance Document on Analytical Parameters for the Determination of Per- and Polyfluoroalkyl Substances (PFAS) in Food and Feed, ANNEX version 1.0 of 11 May 2022. Available online under https://eurl-pops.eu/core-working-groups#_pfas.
- Standard Method Performance Requirements (SMPRs®) for Per- and Polyfluoroalkyl Substances (PFAS) in Produce, Beverages, Dairy Products, Eggs, Seafood, Meat Products, and Feed. AOAC SMPR® 2023.003. https://www.aoac.org/wp-content/uploads/2023/11/SMPR-2023_003.pdf.
- Dreolin N., Foddy H., Organtini K., Adams S., Rosnack K., Hancock P. (2023), Best practices for monitoring PFAS contamination in a routine shared-space commercial laboratory. Waters White Paper. 720007905.
- Organtini K., Oehrle S., Hird S., Adams S., Jandova R. (2021), Matrix Matching or Isotope Dilution? A Comparison of Two Quantitation Approaches to Determine PFAS in Dairy Milk. Waters Application Note. 720007687. August 2022.
- Adams S., Dreolin N., Organtini K., Hancock P. (2023). Analysis of Per- and Polyfluoroalkyl Substances (PFAS) in Animal Products with an Enhanced Sensitivity LC-MS/MS Method using Fish Reference Materials as a Case Study. Waters Application Note. 720008108. October 2023.
- Magnusson B. and Örnemark U. (eds.) Eurachem Guide: The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, (2nd ed. 2014). ISBN 978-91-87461-59-0.
- Theurillat X., Mujahid C., Eriksen B., Griffin A., Savage A., Delatour T., Mottier P. (2023) An LC-MS/MS method for the quantitative determination of 57 per- and polyfluoroalkyl substances at ng/kg levels in different food matrices. Food Additives & Contaminants: Part A, 40:7, 862–877, DOI: 10.1080/19440049.2023.2226771.
Appendix
Featured Products
720008219, February 2024