塩プラグ注入メソッドによる高分子核酸の陰イオン交換分析の改善
要約
核酸ベースの治療薬のパイプラインが急成長する中、さまざまな高分子核酸分子種の濃度、完全性、相対存在量を迅速に確認できる、改善された分析法が必要になっています。特に、陰イオン交換は、mRNA の濃度および完全性を決定するための分析手法としての使用において大きな可能性がありますが、現在の分析法ではキャリーオーバーが多い傾向があり、この手法の導入は現実的ではありません。本研究では、カラムテクノロジーの選択が重要になり得ること、および Gen-Pak™ FAX 弱陰イオン交換カラムが新しい分析法の非常に有効な出発点になり得ることを説明します。さらに、分析種のカラム滞留時間を短くし(1)、最大 100 mM という比較的高い対イオン濃度でグラジエントを開始して(2)、吸引したサンプルを調節塩プラグでブラケットする注入アプローチを適用することで、キャリーオーバーを低減することができます。本研究は、1 本鎖 mRNA の変性とキャリーオーバー、および選択性と分離の関係を解析するのにも役立ちました。移動相の温度を自己フォールディングの溶融温度に近づけると、選択性が増大しますが、キャリーオーバーも増加します。
アプリケーションのメリット
- 連続注入間での低いキャリーオーバー
- mRNA サンプルの回収率の向上
- 併行精度および分析法の頑健性の向上
はじめに
近年、バイオ医薬品業界では遺伝子治療への関心が大幅に増大しています。合成核酸により、新しい形のワクチン接種や、タンパク質発現調節の新しい手段が提供されます。より最近では、in vitro 転写される mRNA が大きな注目を集めています1,2。
インタクト mRNA 原薬を分析するには、液体クロマトグラフィー(LC)手法が必要です3。 多くの場合、イオン対逆相(IP-RP)クロマトグラフィーやサイズ排除(SEC)クロマトグラフィーによる分析スケールの分離が適用され、不均一性の検出や分子の完全性の確認に使用されています3。
オリゴヌクレオチドや核酸の大スケール陰イオン交換(AEX)分離(精製)が通常行われているにもかかわらず、分析スケールの AEX 分離分析法はこれまでわずかしか報告されていません。分析目的の AEX の使用が限られている理由はおそらく、高分子核酸によく見られる低回収率やキャリーオーバーの影響が大きいなど、分析法の頑健性が不足しているためと考えられます。Bridgeneau らは、RNA アプタマー精製における AEX クロマトグラフィー実行の間のキャリーオーバーを防ぐには、特定の条件での 30 分間の平衡化およびブランク分析が必要であると報告しています4。 Guilherme らは、キレート剤および/または変性剤(例:尿素、ホルムアミド、イソプロパノール)を溶出バッファーに補充して分子間相互作用を抑えることでキャリーオーバーを低減する解決策を提案しました5。 多くの電荷を持つ陽イオン分子(スペルミジン)も添加剤として提案されています。これらはコンパクトなスーパーコイル構造を安定させ、回収率を向上させる効果があると考えられています6。 最近、弱イオン対陽イオンのグラジエントによって mRNA の AEX 分離が行えるという興味深い結果が示されました7。 このいわゆるイオン対陰イオン交換「IPAX」分析は、従来の塩グラジエントと比較して、回収率および選択性が向上しています。溶質イオンと固定相リガンドの間で発生する非特異的相互作用に加えて、カラムハードウェアの材質自体も、回収率低下とキャリーオーバー増加の原因になることがあります。現状では、Minkner らは、AEX での siRNA の 95% を超える回収率を優れた性能とみなしており、高分子 mRNA の回収率が小さな mRNA と比べて低いことは、AEX だけでなく、アフィニティークロマトグラフィー分離や疎水性相互作用クロマトグラフィー(HIC)分離でも報告されています8,9。
上記の検討事項を念頭に、改善した分析法を確立できれば、AEX クロマトグラフィー分離が、インタクトな高分子核酸サンプルの分析において信頼できるアプローチになる可能性があると考えられます。今回、AEX 分離で発生するキャリーオーバーを大幅に低減できる効率的な解決策を提案します。
実験方法
サンプルおよび移動相の調製
ClenCap 5 moU EPO mRNA(長さ:858 ヌクレオチド)、ルシフェラーゼ(LUC)mRNA(長さ:1929 ヌクレオチド)、および Cas9 mRNA(長さ:4521 ヌクレオチド)は TriLink Biotechnologies(米国カリフォルニア州サンディエゴ)から購入しました。サンプルを水で 25 µg/mL に希釈し、それ以上の調製を行わずに注入しました。
トリス-(ヒドロキシメチル)アミノメタン(TRIS)、グアニジン塩酸塩(Gdn-HCl)、臭化ナトリウム(NaBr)は Sigma-Aldrich(スイス、Buchs)から購入しました。トリスバッファーは 25 mM 溶液として調製し、pH を約 7.6 に調整しました。この 25 mM トリスバッファーを移動相 A として使用しました。移動相 B には、2 M Gdn-HCl または 2 M NaBr を溶解した 25 mM トリスバッファーを使用しました。
塩プラグ注入では、移動相 B、または 2 M NaBr を含有する 10 倍濃度の BioResolve™ CX B 濃縮液(製品番号:186009064)のいずれかを使用しました。
LC 条件
|
LC システム: |
ACQUITY™ UPLC™ H-Class PLUS Bio システム(クオータナリー) |
|
検出: |
260 nm での UV 検出 |
|
バイアル: |
ポリプロピレンバイアル(製品番号:186002639) |
|
カラム: |
Gen-Pak FAX 陰イオン交換カラム、2.5 µm、4.6 × 100 mm(製品番号:WAT015490) |
|
カラム温度: |
周囲温度 ~ 45 ℃ |
|
サンプル温度: |
5 ℃ |
|
注入量: |
2.0 µL(サンプル) |
|
ブラケット注入シーケンス: |
1.0 µL(塩プレプラグ) + 2.0 µL(サンプル) + 1.0 µL(塩ポストプラグ) |
|
流速: |
0.6 mL/分 |
|
移動相 A: |
25 mM TRIS 水溶液(pH = 7.6) |
|
移動相 B: |
2 M グアニジン塩酸(Gdn-HCl)含有 25 mM TRIS(pH = 7.6)または 2 M 臭化塩素(NaBr)含有 25 mM TRIS(pH = 7.6) |
|
グラジエント: |
高速分離のための推奨の急勾配グラジエント: - 2 M Gdn-HCl 移動相の場合:6 分で 0 ~ 25% B - 2 M NaBr 移動相の場合:7 分で 15 ~ 50% B より高い選択性のための推奨の緩やかなグラジエント: - 2 M NaBr 移動相の場合:15 分で 12 ~ 35% B |
カラムコンディショニング
最低カラム容量の 20 ~ 50 倍の移動相を使ってカラムを平衡化します。次に連続して(3 ~ 4 回)対象サンプルの高ロード量(例:5 ~ 10 µg)注入を行い、固定相の活性部位をコンディショニングします。
結果および考察
生体分子(高分子)で見られる注入の併行精度不良および高キャリーオーバー効果の背景にある現象は多くの場合、表面での望ましくない二次的相互作用、および高分子システムで発生する分子間および分子内の相互作用に関連しています。高分子は全般に表面活性分子であり、さまざまな種類の表面との接触に際して非特異的吸着が起きます10。 このプロセスは、溶質存在量の低下や凝集を引き起こす可能性があるため、非常に重要になる場合があります。吸着時には、ほとんどの高分子で形状の変化(アンフォールディングなどの立体構造変化)が起きます。高分子が占める吸着剤表面の領域はしばしば「フットプリント」と呼ばれます11。 フットプリントは通常、滞留時間とともに増加し、この過程は「拡散プロセス」と呼ばれることがあります。フットプリント関連で生じた余分な吸着の一部は通常、可逆的です12,13。 吸着の速度とフットプリントの表面積は、溶質の濃度にも強く依存します14。 分析種の濃度が高いと、表面の占有が短時間になるため、拡散が生じる時間も短くなります。これにより、平均のフットプリントが小さくなりますが、吸着密度は高くなります。溶媒の pH や塩濃度などの他のパラメーターも、フットプリントのサイズと拡散に影響することがあります。
複数の研究で、上記の効果の一部が実証されています15–18。
滞留時間および初期移動相強度の効果
予備実験により、mRNA が固定相に結合できる時間の長さがキャリーオーバーと相関する可能性が示唆されました。
mRNA は、AEX によって分離される際に、溶出メカニズムのようなオン-オフ(結合と溶出)メカニズムに従うことがすでに確認されています7。 したがって、mRNA はカラムのヘッドに結合し、溶出用のグラジエントの移動相組成になるまで不動になると考えるのが妥当です。そのため、分析種の拡散が発生し、固定相との多点相互作用が形成されるのに要する一定の時間ができます。滞留時間が短くなると結合セグメントが少なくなり、ひいては吸着の相互作用が弱まって、キャリーオーバーが減少するという印象があるかも知れません。
滞留時間がキャリーオーバーに及ぼす影響を確認する体系的実験が実施されています。6 分間で 0 ~ 25% B の Gdn-HCl グラジエントをプログラミングし、グラジエント開始の前に、異なる初期アイソクラティック保持時間(0% B)を設定しました。具体的には、0、0.5、1、2、4、8 分間の初期アイソクラティックホールドを設定しました。EPO および Cas9 の mRNA サンプルを注入し、サンプル注入後のブランク注入におけるキャリーオーバーを(% で)測定しました。図 1A に、生じたキャリーオーバーを溶質の結合時間の関数として示しています。このプロットは、キャリーオーバーと結合時間の間に相関があることを示唆しています。結合時間が短いほど、キャリーオーバーは少なくなっています。予想どおり、これらの実験は、キャリーオーバーを制限する手段として、分析の実行時間を短くする必要があることを示唆しています。保持時間を約 3 分に制限すると、長いグラジエントでは 10 ~ 20% のキャリーオーバーが見られたのに対して、4 ~ 8% とキャリーオーバーが少なくなりました。
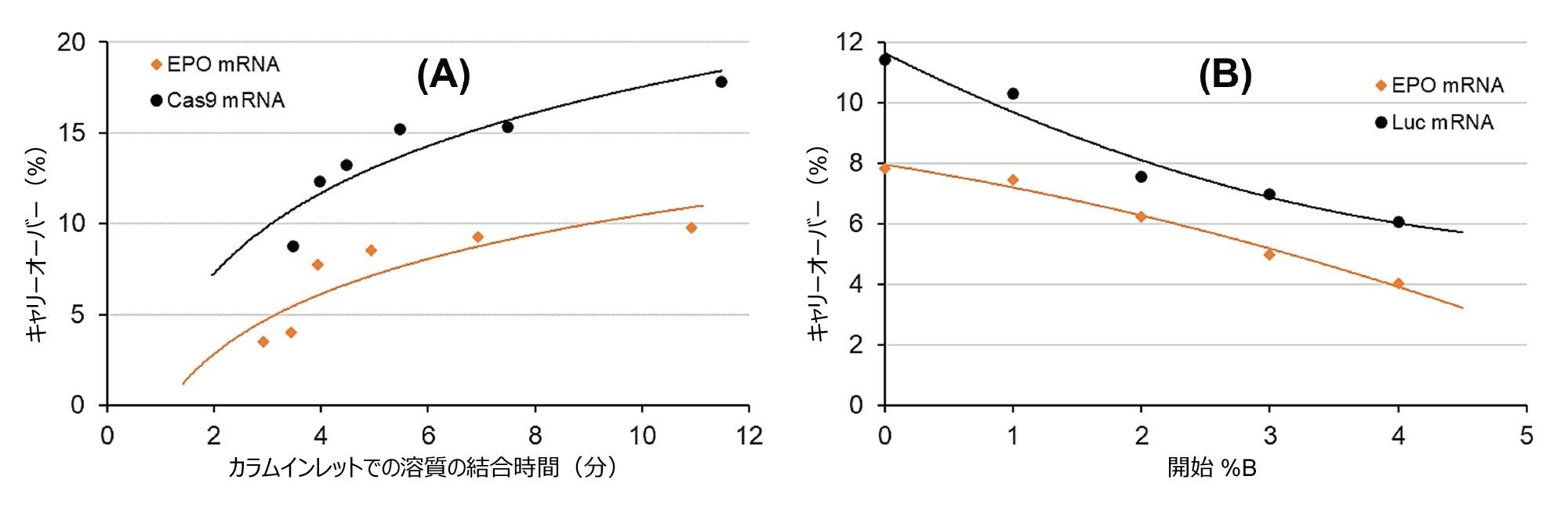 図 1. AEX において mRNA 溶質の結合(滞留)時間(A)および初期移動相強度(B)がキャリーオーバーに及ぼす影響。カラム:Gen-Pak FAX 100 × 4.6 mm、2.5 µm カラム、移動相 A:25 mM TRIS(pH = 7.6)、移動相 B:25 mM TRIS(pH = 7.6) + 2 M Gdn-HCl、F = 0.6 mL/分、グラジエント:6 分で 0 ~ 25% B、周囲温度(約 22 ℃)。
図 1. AEX において mRNA 溶質の結合(滞留)時間(A)および初期移動相強度(B)がキャリーオーバーに及ぼす影響。カラム:Gen-Pak FAX 100 × 4.6 mm、2.5 µm カラム、移動相 A:25 mM TRIS(pH = 7.6)、移動相 B:25 mM TRIS(pH = 7.6) + 2 M Gdn-HCl、F = 0.6 mL/分、グラジエント:6 分で 0 ~ 25% B、周囲温度(約 22 ℃)。
別の実験設定で、移動相の初期強度の影響を試験しました。溶質の拡散が固定相の表面で発生すると仮定した場合、最初の結合時に発生する相互作用が弱いと、拡散はあまり顕著ではない(または遅くなる)とも考えられます。そこで、グラジエント時間を固定し、初期の %B 組成を 0、1、2、3、4% B と変化させました。図 1B に、EPO および Luc mRNA における実測キャリーオーバーを開始時の移動相組成の関数として示しています。開始時の %B が高いほど、キャリーオーバーは少なくなるという明らかな傾向があります。グラジエントを 0% B ではなく 4% B(約 80 mM の対イオン)から開始すると、キャリーオーバーが 2 分の 1 に減少しました。この結果は、0% B ではなく、グラジエントの %B がほどよく高い移動相組成(50 ~ 100 mM の対イオン)から開始する必要があることを示唆しています。
結合時間および開始時 %B の影響はサンプルによって異なるため、目的のサンプル用に最適化する必要があることに注意してください。
塩プラグ(ブラケット注入)でのサンプルプラグの取り囲み
溶媒強度の不一致とは、注入溶媒と移動相の溶離液強度が異なる状況を説明するために使用される用語です19。 溶媒強度の不一致は、サンプル溶媒が移動相組成より強い場合に特に問題になります。このような状況では、部分的または全体的なブレークスルー効果、または少なくともピークのフロンティングやピーク割れが発生することが多くなります19。 このような効果は、多次元分離や親水性相互作用クロマトグラフィー(HILIC)分析で頻繁に発生します。強溶媒の影響を抑えるため、プレサンプルプラグとポストサンプルプラグのセットの希釈によって吸い上げたサンプルをブラケット化する特別にプログラミングされた注入シーケンスを適用して、サンプルをカラムに導入することができます。このような注入シーケンスは、分離性能を向上させるためやブレークスルー効果を抑えるために、すでに適用されています20,21。
結合の相互作用が本質的に非常に強い mRNA の AEX では、上記の注入シーケンスアプローチ(通常は初期結合の相互作用の強度を強めるために適用される)を逆にする必要があります。この場合、初期結合の強度を抑えるために、サンプルを強溶媒プラグとともに注入する必要があります。そこで、高濃度の塩(対イオン)を含む溶媒プラグでサンプルをブラケット化することを提案します。高塩濃度に加えて、溶媒プラグの pH を固定相の官能基の pKa の近く(弱陰イオン交換体の場合は pH 10 ~ 11)になるように調整することもできます。ここでも、この複合効果は、初期吸着の強度を抑えることを目的としています。
シーケンス注入の最も重要な要因、およびそれらが AEX 分離での mRNA のキャリーオーバーにどのように影響するかを確認するために、系統的実験を行いました。試験した要因は、(1)塩プレプラグの容量、(2)塩ポストプラグの容量、(3)ブラケット化に用いる塩プラグの総容量、(4)使用する塩の種類(NaCl、(NH4)2SO4、Gdn-HCl、NaBr)、(5)塩プラグの pH でした。
最も有益なシーケンスは、サンプルプラグを 2 M NaBr 溶液(pH 約 10)のプレプラグおよびポストプラグでブラケット化する注入シーケンスであることがわかりました。図 2 に、ブラケット化注入の概略図を示します。10.5 ~ 11.0 より高い pH では、mRNA が変性し、塩基対形成や塩基スタッキングの相互作用が壊れる可能性があります。高 pH では、インタクト RNA が直鎖状になり、拡散、結合フットプリントの拡大、大幅に強い吸着につながる可能性があります。さらに、一部の核酸塩基は pH 10 を超えると脱プロトン化し、核酸分析種に負電荷が追加されます22。 これが、高 pH 条件で AEX における mRNA の保持が強くなる理由と考えられます3。 したがって、高すぎる pH(>11)は溶媒プラグとして適していません。経験的に、キャリーオーバーの増加が認められます。
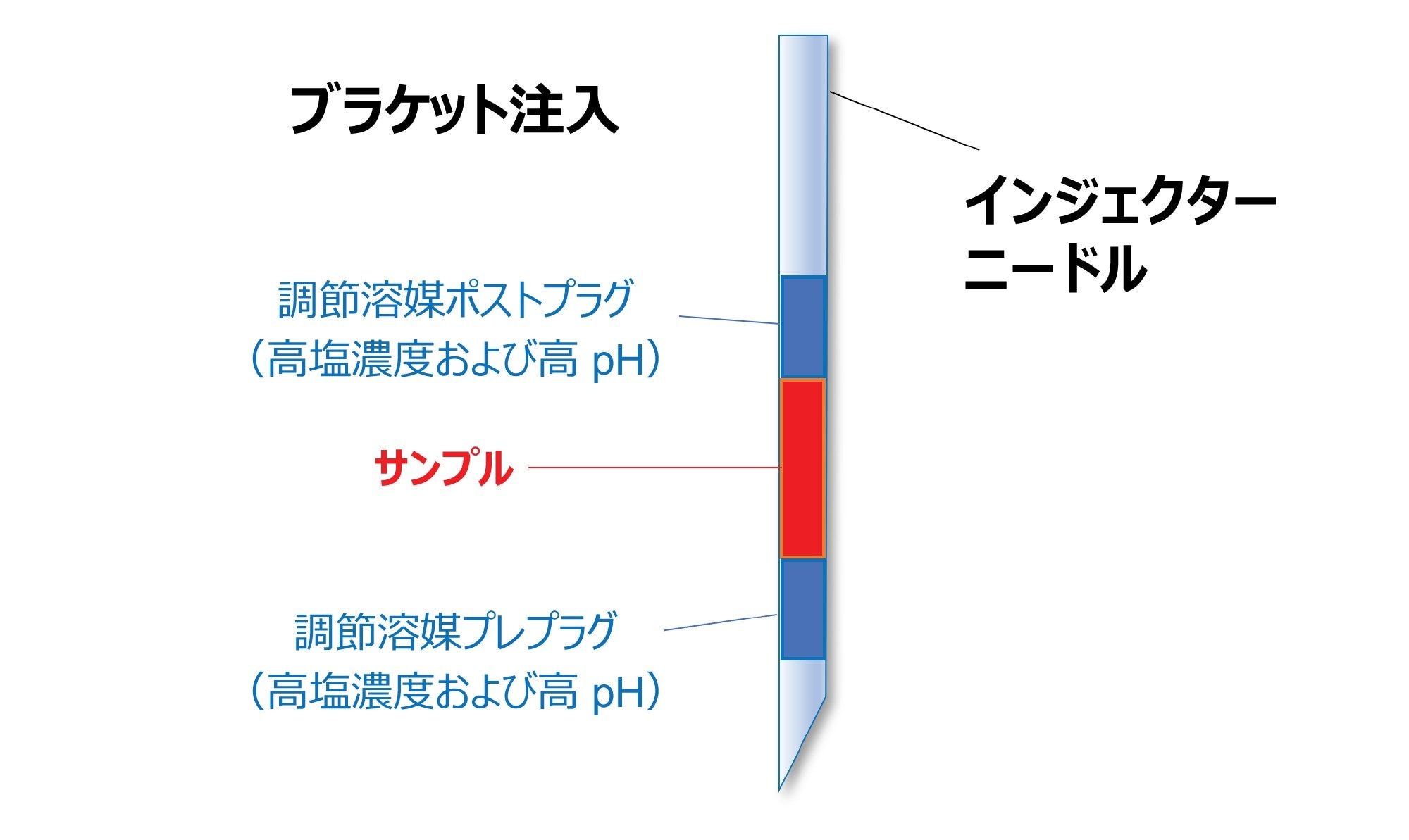 図 2. 吸引したサンプルが高塩濃度および高 pH のモジュレータープラグでブラケット化されている注入シーケンスの概略図。Empower™ ソフトウェアでは、「自動追加」オプションにより、サンプルセットに必要な任意の注入シーケンスを作成することができます23。
図 2. 吸引したサンプルが高塩濃度および高 pH のモジュレータープラグでブラケット化されている注入シーケンスの概略図。Empower™ ソフトウェアでは、「自動追加」オプションにより、サンプルセットに必要な任意の注入シーケンスを作成することができます23。
サンプルのブレークスルーを回避しつつキャリーオーバーが最小になる条件を見つけるには、サンプルプラグの容量に対する調節塩プラグの容量の比を最適化する必要があります。図 3 に、EPO mRNA のキャリーオーバーおよび回収率の変化を、サンプル容量に対する調節プラグ容量の比(Vmod / Vsample)の関数として示しています。これは、プレプラグとポストプラグの容量が同じである場合に使用できます。この図から、(Vmod / Vsample)が約 1.2 ~ 1.3 の値に達するまで、キャリーオーバーが減少して回収率が増加することがわかります。この「限界」値を超えると、注入したサンプル容量の一部が強調節プラグに取り込まれ、部分的なブレークスルーピークがクロマトグラムに表れます。(Vmod / Vsample)≥ 2 の場合、mRNA のピーク全体がカラムのデッドタイムで溶出します(トータルブレークスルー)。(Vmod / Vsample )を約 1 ~ 1.2 に設定すると、塩プラグによる調節なしの場合に見られることが多い 10 ~ 20% のキャリーオーバーではなく、わずか 2 ~ 3% のキャリーオーバーになります。理想的な(Vmod / Vsample)比は、サンプル、インジェクター装置、システム容量、移動相、カラムによって異なることに注意してください。分析法の設定ごとに個別に最適化することが必要です。(Vmod / Vsample)を約 1 に設定するのが出発点として適していると考えられます。例えば、2 µL の mRNA サンプルを注入する場合、適切な出発点は、モジュレータープレプラグ 1 µL + サンプル 2 µL + モジュレーターポストプラグ 1 µL のシーケンスのプログラムになります。
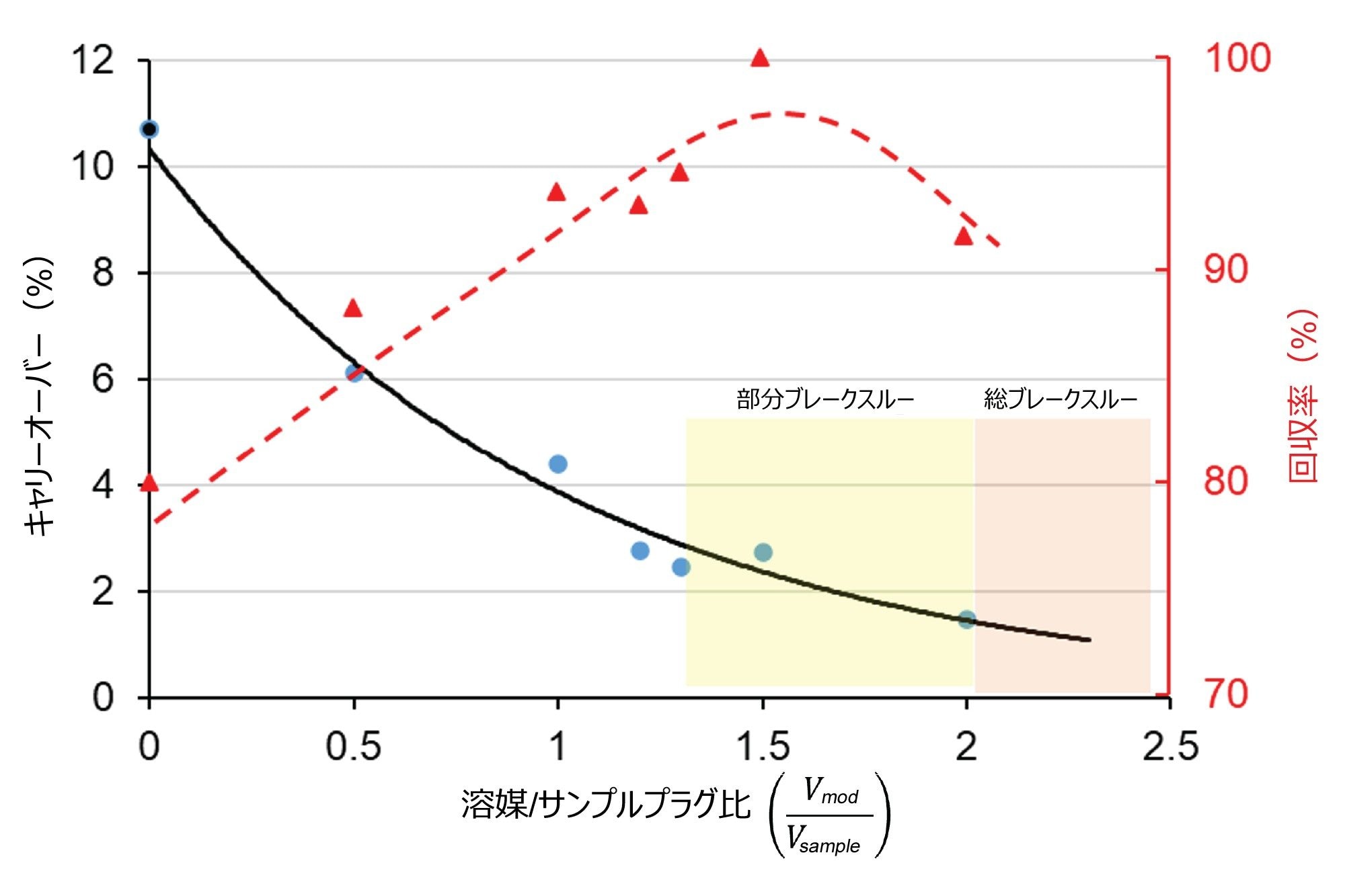 図 3. サンプル容量に対する調節プラグ容量の比がキャリーオーバー(黒色の曲線)および回収率(赤色の曲線)に及ぼす影響。カラム:Gen-Pak FAX 100 × 4.6 mm、2.5 µm カラム、移動相 A:25 mM TRIS(pH = 7.6)、移動相 B:25 mM TRIS(pH = 7.6) + 2 M NaBr、F = 0.6 mL/分、グラジエント:7 分で 15 ~ 50% B、周囲温度(約 22 ℃)。サンプル:EPO mRNA(2 µL 注入)、調節プラグ:移動相 B。
図 3. サンプル容量に対する調節プラグ容量の比がキャリーオーバー(黒色の曲線)および回収率(赤色の曲線)に及ぼす影響。カラム:Gen-Pak FAX 100 × 4.6 mm、2.5 µm カラム、移動相 A:25 mM TRIS(pH = 7.6)、移動相 B:25 mM TRIS(pH = 7.6) + 2 M NaBr、F = 0.6 mL/分、グラジエント:7 分で 15 ~ 50% B、周囲温度(約 22 ℃)。サンプル:EPO mRNA(2 µL 注入)、調節プラグ:移動相 B。
カラム(固定相)によって異なるキャリーオーバー
弱 AEX 相および強 AEX 相の両方を含む、さまざまな固定相を試験しました。若干の違いが見られ、弱交換体では全般にキャリーオーバーがわずかに減っていました。ただし、キャリーオーバーは常にサンプルおよび条件によって異なることに注意してください。弱イオン交換体の間でも差が見られることがありますが、これはおそらくリガンドの密度やリガンドのアクセス性(形態)の差、および水素結合などのその他の相互作用に関連していると思われます。
図 4 に、Gen-Pak FAX 弱陰イオン交換カラムと弱陰イオン交換モノリスカラムの比較を、両方のカラムに最適化した同じブラケット注入を使用して示しています。Gen-Pak FAX カラムでは、キャリーオーバーが顕著に少なく分離度は同等であり、ピークの鋭さが大幅に向上している場合がありました。
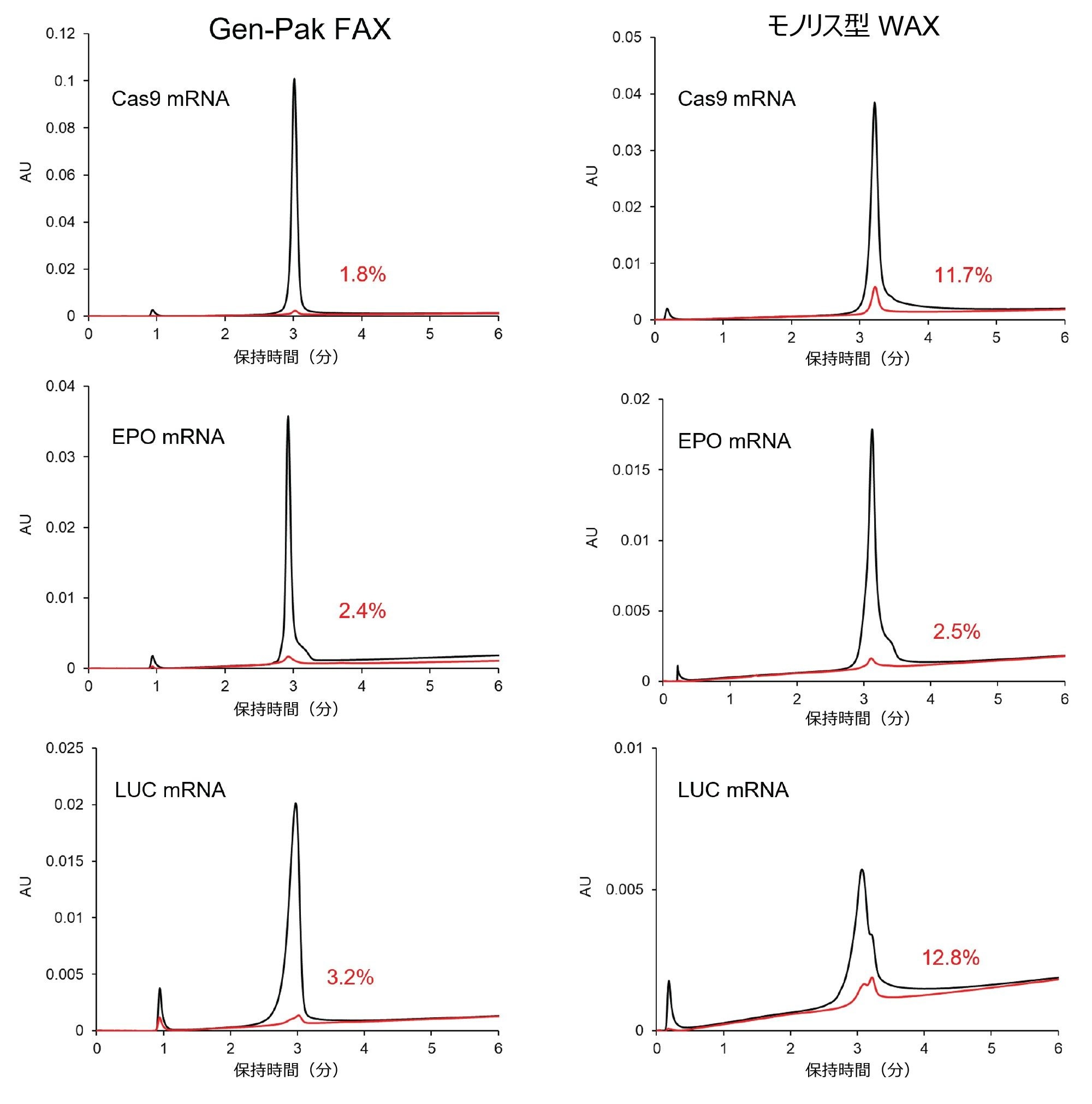 図 4. 2 種類のカラムで見られたキャリーオーバーの比較。カラム:Gen-Pak FAX 100 × 4.6 mm、2.5 µm カラム(左)、およびモノリス型 WAX 4.95 × 5.2 mm(右)。グラジエント条件:図 3 と同じ、調節溶媒プラグ:2 M NaBr 含有 pH 10.2 バッファー。ブラケット注入:調節プレプラグ 1 µL + サンプル 2 µL + 調節ポストプラグ 1 µL。% で表されている赤色の数字は、サンプル注入後のブランク注入で見られたキャリーオーバーの割合に対応します。
図 4. 2 種類のカラムで見られたキャリーオーバーの比較。カラム:Gen-Pak FAX 100 × 4.6 mm、2.5 µm カラム(左)、およびモノリス型 WAX 4.95 × 5.2 mm(右)。グラジエント条件:図 3 と同じ、調節溶媒プラグ:2 M NaBr 含有 pH 10.2 バッファー。ブラケット注入:調節プレプラグ 1 µL + サンプル 2 µL + 調節ポストプラグ 1 µL。% で表されている赤色の数字は、サンプル注入後のブランク注入で見られたキャリーオーバーの割合に対応します。
温度の影響
AEX クロマトグラフィーでは、温度を上げると、RNA サンプルのピークが少なくなり、より明確になることが報告されています24。 これは、二次構造の減少によるものと考えられています。さらに、高温では保持の増大も観察されましたが、これも自己構造の減少によって説明できます。最近、温度も AEX における mRNA の回収率とキャリーオーバーに大きな影響を及ぼすことがわかりました7。 多くのパラメーターが mRNA、水系移動相、固定相の間の相互作用の強さを変化させる可能性があるため、温度の影響によるキャリーオーバーの変化を予測することは困難です。大量の塩の存在下では、mRNA の疎溶媒性効果、塩析、塩溶、脱水、および構造的再配列のすべてが、多かれ少なかれ発生する可能性があります7。 そのため、ブラケット化注入に対する温度の影響についての試験に関心を向けました。図 5 に、EPO および Cas9 の mRNA サンプルを周囲温度および高温(T = 35 ℃ および 45 ℃)で注入して得られたクロマトグラムを示します。キャリーオーバーが温度とともに増大するように見えますが、これは保持の増大が観測されることと一致しています(高温では結合が強いことを示唆しています)。したがって、サンプルのキャリーオーバーを検討する場合は、周囲温度で実験することが推奨されます。一方、ピーク形状、選択性、分離プロファイルが温度によって大きく変化することから、サンプル成分の分離に対して、対応する有利な効果があるように思われます。最終的には、2 つの分析法(周囲温度条件での分析、および高いカラム温度での分析)を設定することが有用と考えられます。周囲温度(低温)を使用する分析法は存在量/濃度を決定する測定に適し、高温を使用する分析法は mRNA の生物物理学的特性や化学的不均一性の調査に有用である可能性があります。
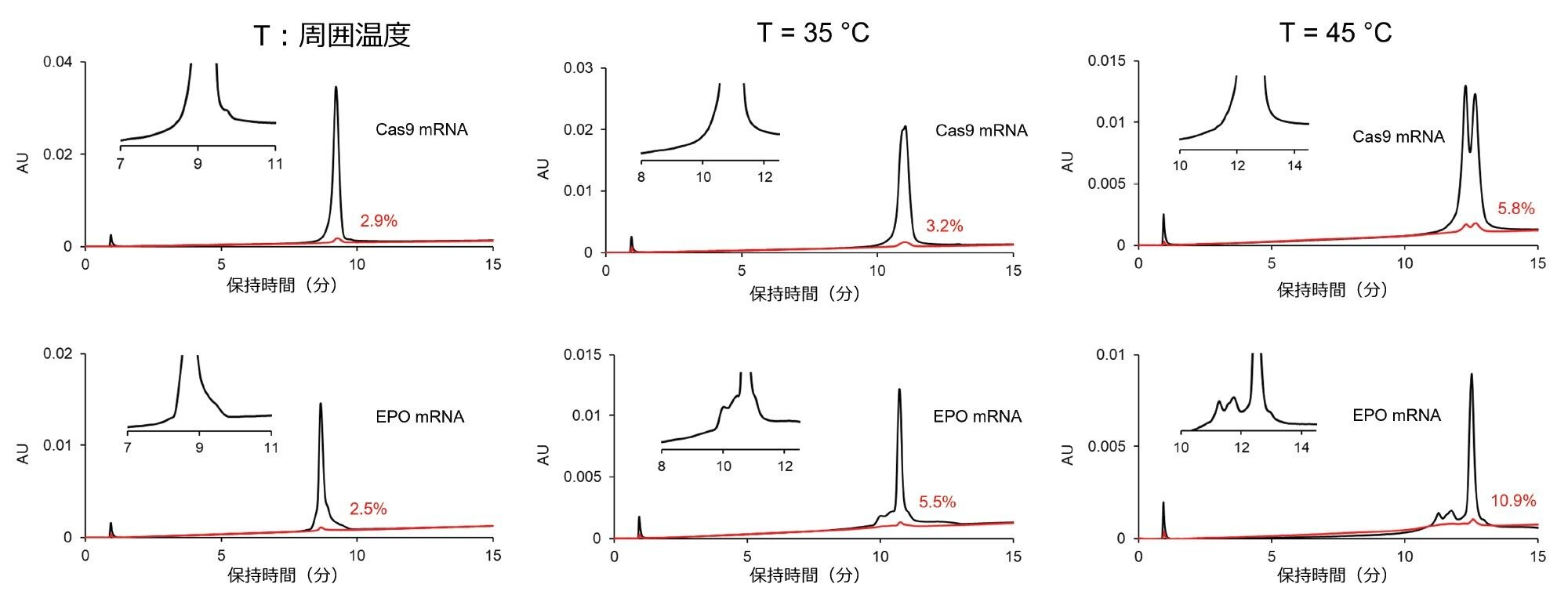 図 5. クロマトグラフィープロファイル(選択性)およびキャリーオーバーに対する温度の影響。カラム:Gen-Pak FAX 100 × 4.6 mm、2.5 µm カラム、移動相 A:25 mM TRIS(pH = 7.6)、移動相 B:25 mM TRIS(pH = 7.6) + 2 M NaBr、F = 0.6 mL/分、グラジエント:15 分間で 12 ~ 35% B(緩やかなグラジエント)、調節溶媒プラグ:移動相 B。ブラケット注入:調節プレプラグ 1 µL + サンプル 2 µL + 調節ポストプラグ 1 µL。温度:周囲温度(左)、35 ℃(中央)、45 ℃(右)。
図 5. クロマトグラフィープロファイル(選択性)およびキャリーオーバーに対する温度の影響。カラム:Gen-Pak FAX 100 × 4.6 mm、2.5 µm カラム、移動相 A:25 mM TRIS(pH = 7.6)、移動相 B:25 mM TRIS(pH = 7.6) + 2 M NaBr、F = 0.6 mL/分、グラジエント:15 分間で 12 ~ 35% B(緩やかなグラジエント)、調節溶媒プラグ:移動相 B。ブラケット注入:調節プレプラグ 1 µL + サンプル 2 µL + 調節ポストプラグ 1 µL。温度:周囲温度(左)、35 ℃(中央)、45 ℃(右)。
結論
高分子核酸の陰イオン交換分析では、キャリーオーバーが多いことと、回収率が低いことが知られています。今回、キャリーオーバーを低減して分析法の頑健性を向上させるための、新しい分析法に関する検討事項を提案します。過去の複数回の調査を通じて、Gen-Pak FAX カラムにより、高分子 1 本鎖核酸サンプルが最も効果的に分離されることが確認されています。このカラムは、新しい分析法を導入するための信頼できる出発点になります。
キャリーオーバーに関しては、分析時間を短くすることが好ましいことが分かっています。比較的強い溶離液強度でグラジエントを開始することも役立ちます。
しかし、最も有用なアプローチは、サンプルをいわゆる「調節」プラグでブラケット化する特別にプログラミングされたサンプル注入を適用することです。これらのプラグには高濃度の塩が含まれており、約 pH 10 になるように緩衝化されています。このブラケット注入により、カラムヘッドでの溶質の結合強度が弱まり、回収率とキャリーオーバーの点で改善します。調節プラグの容量および比は、個々の分析法のセットアップごとに最適化する必要があります。この試験では、(Vmod / Vsample)比約 1 がかなり有効であることがわかりました。Gen-Pak FAX カラムでこの新規のブラケット注入モードを使用することにより、高分子 mRNA のキャリーオーバーが、従来の AEX 分析法では多くの場合 10 ~ 20% であるのに対して、約 2% に低減することができました。
参考文献
- C. Rinoldi, S.S. Zargarian, P. Nakielski, X. Li, A. Liguori, F. Petronella, D. Presutti, Q. Wang, M. Costantini, L. De Sio, C. Gualandi, B. Ding, F. Pierini.Nanotechnology-assisted RNA delivery: from nucleic acid therapeutics to COVID-19 vaccines. Small Methods 5 (2021) 2100402, https://doi.org/10.1002/smtd.202100402
- A. Akinc, M.A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M.J. Hope, T.D. Madden, B.L. Mui, S.C. Semple, Y.K. Tam, M. Ciufolini, D. Witzigmann, J.A. Kulkarni, R. van der Meel, P.R. Cullis.The Onpattro story and the clinical translation of nanomedicines containing nucleic acid based drugs. Nat. Nanotechnol.2019, 14, 1084–1087.https://doi.org/10.1038/s41565-019-0591-y
- S. Fekete, C. Doneanu, B. Addepalli, M. Gaye, J. Nguyen, B. Alden, R. Birdsall, D. Han, G. Isaac, M. Lauber.Challenges and emerging trends in liquid chromatography-based analyses of mRNA pharmaceuticals. J. Pharm. Biomed.Alna., 224 (2023) 115174. https://doi.org/10.1016/j.jpba.2022.115174
- P. Bridonneaua, S. Bunch, R. Tengler, K. Hill, J. Carter, W. Pieken, D. Tinnermeier, R. Lehrman, D.W. Drolet.Purification of a highly modified RNA-aptamer: Effect of complete denaturation during chromatography on product recovery and specific activity. J. Chromatogr. B, 726 (1999) 237–247.
- G N Ferreira, G A Monteiro, D M Prazeres, J M Cabral.Downstream processing of plasmid DNA for gene therapy and DNA vaccine applications. Trends Biotechnol. 18 (2000) 380–388.https://doi.org/10.1016/s0167-7799(00)01475-x
- J.C. Murphy, J.A. Wibbenmeyer, G.E. Fox, R.C. Willson.Purification of plasmid DNA using selective precipitation by compactation agents. Nat. Biotechnol. 17 (1999) 822–823.https://doi.org/10.1038/11777
- S. Fekete, H. Yang, K. Wyndham, M. Lauber.Salt gradient and ion-pair mediated anion exchange of intact messenger ribonucleic acids. J. Chromatogr. Open, 2 (2022) 100031.https://doi.org/10.1016/j.jcoa.2022.100031
- R. Minkner, J. Boonyakida, E.Y. Park, H. Wätzig.Oligonucleotide separation techniques for purification and analysis: What can we learn for today’s tasks?. Electrophoresis, 43 (2022) 2402–2427.https://doi.org/10.1002/elps.202200079
- T. Cui, K. Fakhfakh, H. Turney, G. Güler-Gane, A. Toloczko, M. Hulley, R. Turner.Comprehensive studies on building a scalable downstream process for mRNAs to enable mRNA therapeutics. Biotechnol. Prog.39 (2023) e3301.https://doi.org/10.1002/btpr.3301
- K. Höger, T. Becherer, W. Qiang, R. Haag, W. Friess, S. Küchler.Polyglycerol coatings of glass vials for protein resistance.Eur.J. Pharm.Biopharm. 85(2013) 756–764.https://doi.org/10.1016/j.ejpb.2013.04.005
- M Rodriguez-Allera, A. Cusumano, A. Beck, D. Guillarme, S. Fekete.Importance of vial shape and type on the reproducibility of size exclusion chromatography measurement of monoclonal antibodies. J. Chromatogr. B, 1032 (2016) 131–138.http://dx.doi.org/10.1016/j.jchromb.2016.04.032
- J. Mathes, Protein Adsorption to Vial Surfaces—Quantification, Structural andMechanistic Studies, Ph.D. thesis, Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität, München, 2010.
- J.M. Kleijn, W. Norde.The adsorption of proteins from aqueous solution on solid surfaces. Het. Chem.Rev. 2 (1995) 157–172.
- M.V.D. Veen, M.C. Stuart, W. Norde, Spreading of proteins and its effect on adsorption and desorption kinetics. Colloids Surf. B 54 (2007) 136–142.https://doi.org/10.1016/j.colsurfb.2006.08.017
- S. Fekete, A. Murisier, D. Guillarme.Hydrophobic Interaction Chromatography (HIC) for the Characterization of Therapeutic Monoclonal Antibodies and Related Products, Part 1: Theoretical Aspects. LCGC Europe, 34 [2021] 101–105.
- E. Haimer, A. Tscheliessnig, R. Hahn, A. Jungbauer.Hydrophobic interaction chromatography of proteins IV: Kinetics of protein spreading. J. Chromatogr. A.1139 (2007) 84–94.https://doi.org/10.1016/j.chroma.2006.11.003
- R. Ueberbacher, E. Haimer, R. Hahn, A. Jungbauer.Hydrophobic interaction chromatography of proteins V. Quantitative assessment of conformational changes. J. Chromatogr. A.1198–1199 (2008) 154–163.https://doi.org/10.1016/j.chroma.2008.05.062
- J.A. Queiroz, C.T. Tomaz, J.M.S. Cabral.Hydrophobic interaction chromatography of proteins. J. Biotechnol. 87 (2001) 143–159.https://doi.org/10.1016/S0168-1656(01)00237-1
- S. Chapel, S. Heinisch.Strategies to circumvent the solvent strength mismatch problem in online comprehensive two-dimensional liquid chromatography. J. Sep. Sci., 45 (2022) 7–26. https://doi.org/10.1002/jssc.202100534
- A. C. Sanchez, J.A. Anspach, T. Farkas.Performance optimizing injection sequence for minimizing injection band broadening contributions in high efficiency liquid chromatographic separations. J. Chromatogr. A, 1228 (2012) 338–348.https://doi.org/10.1016/j.chroma.2012.01.038
- M. Taylor, J. Kawakami, D.V. McCalley.Managing sample introduction problems in hydrophilic interaction liquid chromatography. J. Chromatogr. A, 1700 (2023) 464006.https://doi.org/10.1016/j.chroma.2023.464006
- S. Ganguly, K.K. Kundu.Protonation/deprotonation energetics of uracil, thymine, and cytosine in water from e.m.f./spectrophotometric measurements.Can. J. Chem.72 (1994) 1120–1126.
- https://support.waters.com/KB_Inf/Empower_Tips_of_the_Week/WKB121702_In-Needle_Auto_Additions_in_Empower
- A. Kanavarioti.HPLC methods for purity evaluation of man-made single-stranded RNAs. Sci Rep 9 (2019) 1–13. https://doi.org10.1038/s41598-018-37642-z.
720007991JA、2023 年 7 月