酢酸アンモニウム移動相を用いたモノクローナル抗体の迅速 SEC-UV 分析
要約
モノクローナル抗体(mAb)サンプルの凝集を分析するための、酢酸アンモニウム移動相を用いた UV 吸光度検出を使用する 4 分間での高分離能サイズ排除(SEC-UV)分析法を紹介します。迅速非変性 SEC-UV 分析法は、バイオ医薬品における細胞培養、精製、製剤開発の支援において有用です。さらに、酢酸アンモニウム(AMA)移動相を使用する迅速 SEC-UV 分析法も、非揮発性の塩およびバッファー(NaCl、リン酸など)で構成される従来のサイズ排除クロマトグラフィー(SEC)移動相を使用するよりも、LC-MS 分析専用の LC システムにより効率的に導入できます。SEC 分析法には ESI-MS 検出も使用できますが、AMA 濃度と流速が高いため、MS データの質が低下します。
このアプリケーションでは、SEC を効果的に使用して、プロテイン A 精製 mAb サンプル中の、製品の安全性と有効性に影響する可能性のある自己会合または凝集した mAb 不純物をモニターしました。プロテイン A ベースの mAb 精製の自動化を、Andrew+™ ピペッティングロボットを使用して実現させました。ハイスループット SEC-UV 分析法では、ACQUITY™ Premier Protein SEC(250 Å、1.7 µm、4.6 × 150 mm)カラム、流速 0.50 mL/分の 200 mM 酢酸アンモニウム移動相を使用し、UV 吸光度検出(280 nm)を行いました。分析法の性能と頑健性を、BioAccord™ LC-MS(ESI-Tof)システムに加えて、スタンドアローンの ACQUITY Premier UPLC™ システムで評価しました。
アプリケーションのメリット
- プロテイン A 精製細胞培養サンプル、アップストリームの精製サンプル、製剤安定性試験用サンプル中の mAb の HMW および LMW サイズバリアントの 4 分間迅速 SEC-UV 分析
- 濃度 0.5 mg/mL 以上で UPLC-MS システムにも適合する、mAb サンプルの効果的な SEC-UV 分析
- 3 か月間にわたって 700 回を超える分析に実使用したカラムの寿命の実証
はじめに
治療用タンパク質の製品の安全性と有効性に影響する可能性がある、自己会合または凝集した mAb 不純物のモニタリングには、SEC が頻繁に使用されています1。 以前の研究で、Waters ACQUITY Premier Protein SEC 250 Å、1.7 µm カラムを用いることで、迅速でハイスループット(HT)のサイズ排除クロマトグラフィー(SEC)分析機能により、低イオン強度で頑健で高分離能の分離が行えることが実証されています2,3。 この試験では、これらの機能が、LC-MS 装置に適合する酢酸アンモニウム(AMA)移動相を使用して、プロテイン A 精製 mAb サンプルの HT SEC-UV 分析の開発に適用されました。
AMA はネイティブ SEC-MS 分析で汎用的に使用されていますが、私たちはこれを SEC-UV 分析における移動相の塩として選択しました。その理由は、リン酸ナトリウムや塩化ナトリウムなどの従来の SECバッファー成分と比較して、LC-MS 装置との適合性が大幅に優れているためです。さらに、AMA は高濃度では静菌性があることが報告されています4。 このことは、AMA について特に評価されていませんが、微生物汚染が起こりやすいリン酸やその他の生物学的バッファーシステムと比較して有益である可能性があります。
さらに、プロテイン A 精製メソッドの自動化に焦点を当てた並行試験の一環として、3 か月以上にわたって行われた 700 回を超える分析について得られたカラム寿命のデータも、カラム間の再現性の結果とともに紹介します。
実験方法
サンプルの説明
mAb は、さまざまなソースから入手し、リン酸バッファー生理食塩水(PBS)で表示濃度に希釈しました。これらには、インフリキシマブ、リツキシマブ、ナタリズマブ、セツキシマブ、トシリズマブ、NISTmAb RM-8671、そして 2 サンプルのトラスツズマブ、トラスツズマブ(Herceptin™)、トラスツズマブ-anns(Kanjinti™)が含まれていました。
LC 条件
|
LC システム: |
ACQUITY Premier UPLC(バイナリーまたはクオータナリーソルベントマネージャー(BSM または QSM)と CH-A カラムヒーターを搭載)または BioAccord™ LC-MS(ESI-ToF)システム |
|
検出: |
ACQUITY UPLC TUV 検出器(5 mm チタンフローセル付き)、 |
|
波長:280 nm |
|
|
バイアル: |
ポリプロピレン 12 × 32 mm スクリューネックバイアル(キャップ付きおよびスリット入り PTFE/シリコーンセプタム付き)、容量 300 µL、100 個入り(製品番号:186002639) |
|
カラム: |
ACQUITY Premier Protein SEC 250 Å、1.7 µm、4.6 × 150 mm カラム + mAb サイズバリアント標準試料(製品番号:176004783) |
|
カラム温度: |
25 ˚C |
|
サンプル温度: |
6 ˚C |
|
注入量: |
5 µL または指示どおり |
|
流速: |
0.5 mL/分 |
|
移動相: |
酢酸アンモニウム、LC-MS グレード(Supelco LiChropur™、LC-MS 用溶離液添加剤、73594)、0.1 µm 滅菌フィルターでろ過、200 mM または指示どおり |
データ管理
|
クロマトグラフィーソフトウェア: |
Empower™ 3(FR 4)および UNIFI™ バージョン 2.1.2.14 |
結果および考察
分析法開発
この試験の目的は、LC-MS 分析専用の UPLC システムに容易に導入できる、mAb 凝集体の分析向けの HT 非変性 SEC-UV 分析法を開発することでした。200 mM の AMA 移動相を使用することでこの目標が達成され、試験した 7 種類の mAb すべてで効果的な分離が得られました。流速 0.5 mL/分を選択したため、分析時間は 4 分になりました。
昇華点が 120 ℃ で、LC-MS 向けに高純度のものを入手できる AMA を、揮発性で MS に対応できる塩として選択しました。AMA は、滴定せずに pH 約 7.0 で使用しました。この pH では緩衝能がほとんどありませんが、酢酸(pKa 4.75)は pH 6.0 未満で、アンモニウム(pKa 9.25)は pH 8.0 超で、それぞれ最大値の 5% 以上の緩衝能を発揮すると予測されます。一連の mAb について、100 mM ~ 300 mM の AMA 濃度を評価しました(図 1)。mAb サンプルは、元の表示濃度に基づいて、PBS で 1.0 mg/mL に希釈しました。HMWS の相対存在量が約 1% またはそれ以下の mAb サンプルには注入量 5 µL を使用し、HMW レベルがそれより高い mAb サンプルには、各 mAb で同程度の HMWS のロード量にするために、より少ない注入量を使用しました。
150 mM 以上の AMA 濃度では、評価したすべての mAb において、同等のプロファイルと HMWS のピーク面積が見られました。一方、100 mM AMA 移動相では、HMWS フォームのピーク面積の減少が両方のトラスツズマブサンプルで認められました。トラスツズマブでは HMWS2 の減少が最も顕著で、HMWS のピーク面積の減少は、程度は低いものの、トシリズマブおよび NISTmAb でも見られました。これらの結果に基づいて、試験したすべての mAb について頑健な分離が得られる 200 mM AMA を最適な移動相として選択しました。低分子量の mAb フラグメント(LMWS1 および LMWS2)も観察されました。大部分の mAb サンプルにおいて、微量の LMWS が存在して分離が困難でしたが(LMWS1 が 0.7 % 未満、LMWS2 が 0.1 % 未満)、150 mM ~ 250 mM AMA で一貫した分離が得られました。
流速 0.5 mL/分を選択したため、分析時間は 4 分になりました。この流速で有効な分離が達成され、さらに、LC-MS システムに導入した場合に MS ダイバートバルブへの接続によって発生する UV フローセルでの過度の圧力が回避されました。
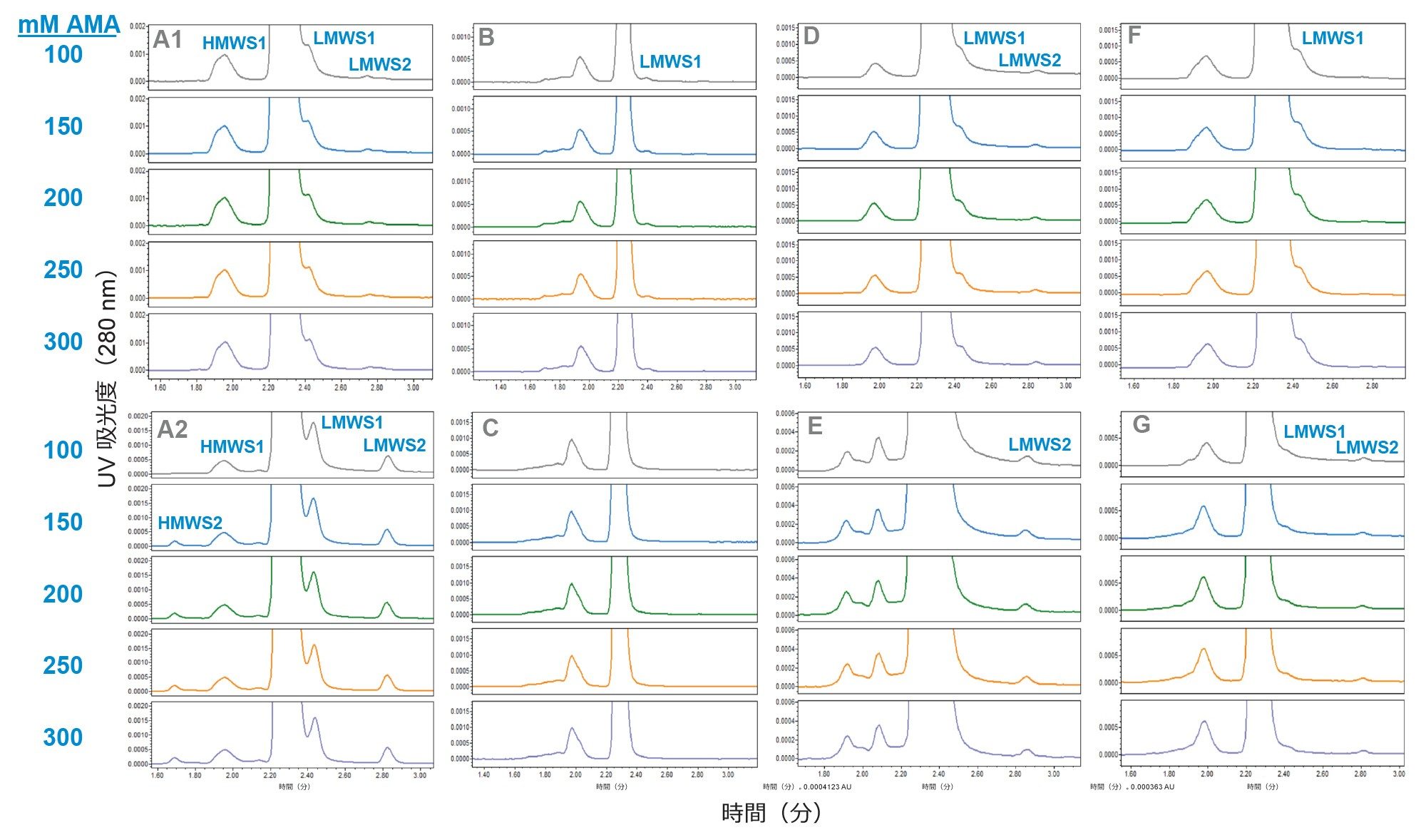 図 1. いくつかの mAb サンプルについて濃度 100 mM ~ 300 mM の AMA 移動相を使用して得られた SEC-UV クロマトグラムを示します。特に記載されていない限り、注入量は 5 µL でした。サンプルは、(A1)トラスツズマブ-anns、(A2)トラスツズマブ、(B)セツキシマブ(0.5 µL)、(C)ナタリズマブ(1 µL)、(D)トシリズマブ、(E)リツキシマブ、(F)インフリキシマブ、(G)NISTmAb RM-8671(1.5 µL)でした。分析した調製物はすべて有効期限を過ぎていました。データは ACQUITY Premier QSM UPLC で収集しました。
図 1. いくつかの mAb サンプルについて濃度 100 mM ~ 300 mM の AMA 移動相を使用して得られた SEC-UV クロマトグラムを示します。特に記載されていない限り、注入量は 5 µL でした。サンプルは、(A1)トラスツズマブ-anns、(A2)トラスツズマブ、(B)セツキシマブ(0.5 µL)、(C)ナタリズマブ(1 µL)、(D)トシリズマブ、(E)リツキシマブ、(F)インフリキシマブ、(G)NISTmAb RM-8671(1.5 µL)でした。分析した調製物はすべて有効期限を過ぎていました。データは ACQUITY Premier QSM UPLC で収集しました。
分析法の評価
サンプル濃度 0.5 ~ 2.0 µg/µL の mAb で、HMWS サイズバリアントおよび LMWS サイズバリアントについて一貫した相対ピーク面積が観察されました。サンプル濃度の影響を評価するために、濃度 0.5、1.0、2.0 µg/µL のトラスツズマブを評価しました。結果を図 2 に示します。3 種類の HMWS サイズバリアント(HMWS2、HMWS1、HMWSs)および 2 種類の LMWS サイズバリアント(LMWS1 および LMWS2)について、クロマトグラフィープロファイルで一貫した相対ピーク面積が観察されました。HMWS1 と HMWS2 は主に二量体および多量体の自己会合 mAb です。HMWSs として同定されたピークは確定的な同定には至りませんでしたが、LMWS mAb フラグメントが含まれる自己会合の結果として生じたものと疑われます。LMWS1 は主に Fab ドメイン(約 100 KDa)が 1 つ欠落した mAb であり、LMWS2 は主に Fab と Fc の混合物(約 50 KDa)です。
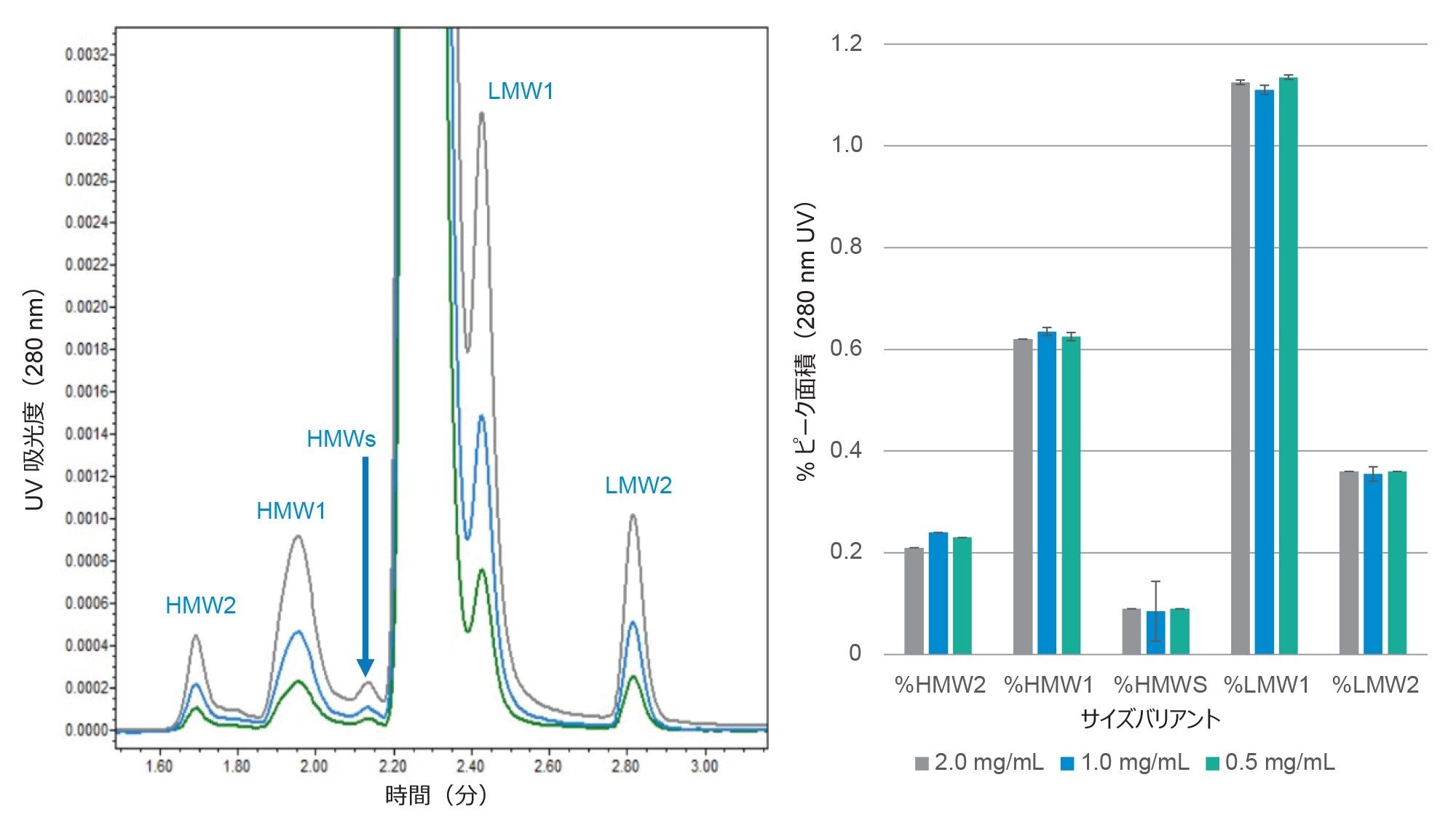 図 2. 濃度 0.5、1.0、2.0 mg/mL で評価したトラスツズマブの SEC-UV クロマトグラムと定量結果を示します。エラーバーは得られた値の範囲を表します(n=3)。注入量は 5 µL で、分析した調製物はすべて有効期限を過ぎていました。データは ACQUITY Premier QSM UPLC で収集しました。
図 2. 濃度 0.5、1.0、2.0 mg/mL で評価したトラスツズマブの SEC-UV クロマトグラムと定量結果を示します。エラーバーは得られた値の範囲を表します(n=3)。注入量は 5 µL で、分析した調製物はすべて有効期限を過ぎていました。データは ACQUITY Premier QSM UPLC で収集しました。
この試験での注入量は 5 µL です。この試験では詳細に調査していませんが、注入量を増やす、より高感度の波長(例:214 nm)の UV 吸光度を使用する、内因性のタンパク質蛍光の検出を使用する、などにより検出下限を下げることができます。
サイズバリアントの相対的定量におけるこの分析法の有効性は、2 種類のトラスツズマブサンプルの混合物を使用しても確認されました(図 3)。この評価では、図 1 に示す 2 種類のトラスツズマブ調製物(トラスツズマブ-anns およびトラスツズマブ)を、個別および 1:1(v:v)混合液として分析しました。使用した主に 80 ℃ で約 10 年間保存されていたトラスツズマブサンプルは、サンプル原液の SEC 分析に基づいて、トラスツズマブ-anns サンプルよりも高い合計 mAb 濃度を示しました。結果として、この 1:1(v:v)混合液は、重量ベースで 56.1% のトラスツズマブ、43.9% のトラスツズマブ-anns から成ると判定されました。HMWS2、HMWS1、LMWS1、および LMWS2 のサイズバリアントについて、1% を下回るまたは 1% に近い相対レベルの機能応答曲線が得られました。
この試験では、部分的に分離したピークにベースラインまで垂線を引く波形解析を使用しました(図 3)。この方法は一般に、はるかに存在量が多い(100 倍大きい)先行するメイン(モノマー)ピークから部分的に分離した LMW1 などのピークの割合を過大評価しますが、ベースラインまで垂線を引く方法により通常、信頼性の高い自動積分アルゴリズムがより容易になる傾向があります。ただし、これは 2 つのピークの間の谷が検出できる場合のみです。この谷が存在するかどうかは、クロマトグラフィー分離の質により異なり、さまざまな変数のうちでも特に、移動相の有効性、カラムの分離能、LC のバンド拡散、および低レベルの成分の相対存在量の影響を受けます5。 これらの試験では、5 σ UPLC のバンド拡散は 12 µL を下回っていました。例えば、この分析法では、LMW1 バリアントの定量値は、トラスツズマブ-anns サンプルで見られたレベル(0.6%)を下回らないと予想されます。
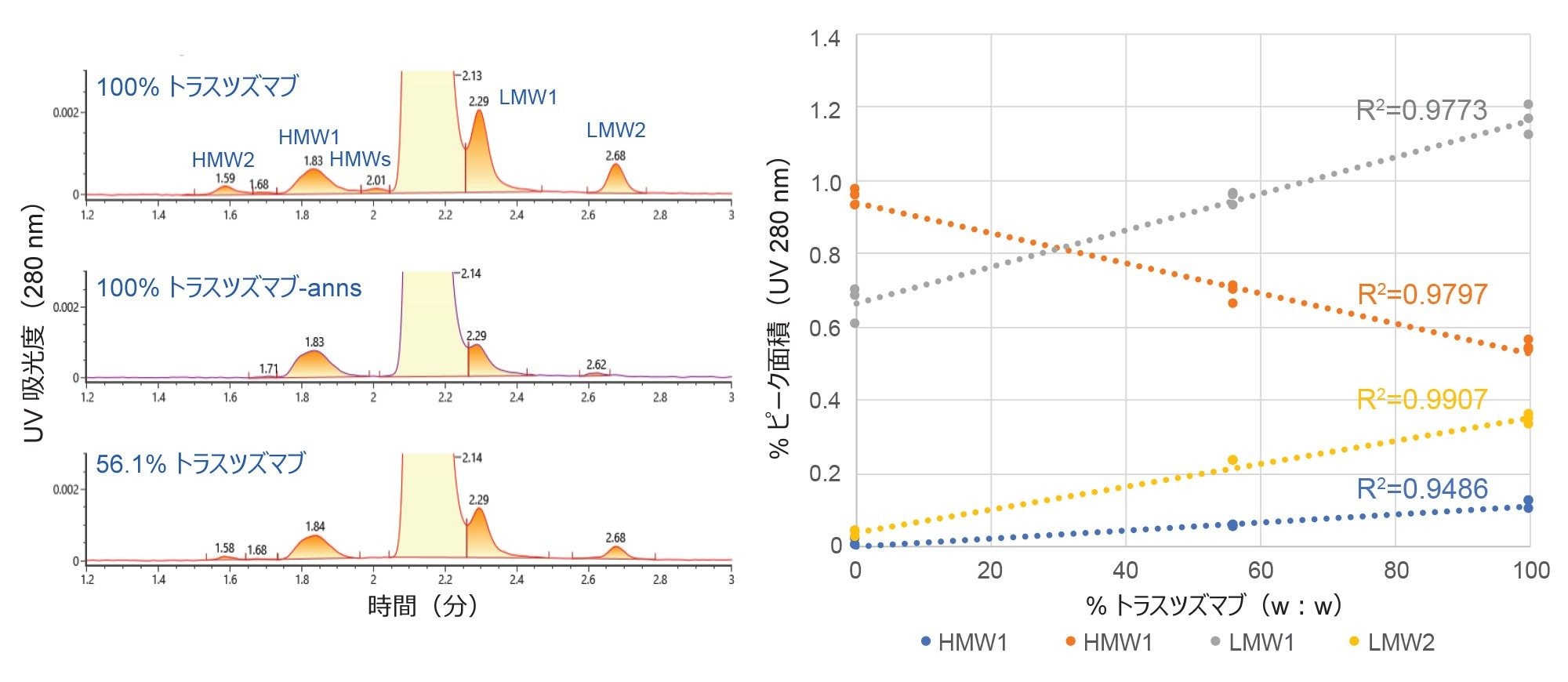 図 3. トラスツズマブ、トラスツズマブ-anns、およびこれら 2 サンプルの混合物の SEC-UV クロマトグラムと定量結果を示します。注入量は 5 µL で、分析した調製物はすべて有効期限を過ぎていました。これらの UV データは BioAccord LC-MS(ESI-Tof)システムで収集したものです。クロマトグラフィーの条件は本文に記載しています。
図 3. トラスツズマブ、トラスツズマブ-anns、およびこれら 2 サンプルの混合物の SEC-UV クロマトグラムと定量結果を示します。注入量は 5 µL で、分析した調製物はすべて有効期限を過ぎていました。これらの UV データは BioAccord LC-MS(ESI-Tof)システムで収集したものです。クロマトグラフィーの条件は本文に記載しています。
この HT 分析法の主な用途の 1 つである、プロテイン A アフィニティー精製 mAb サンプルの試験も実証されました。この評価のため、トラスツズマブ-anns のサンプルを PBS 中で 1 µg/µL になるように調製し、清澄化した遺伝子導入していない CHO 細胞培地(NTM)に 1 µg/µL になるようにスパイクしました。NTM は Syd Labs, Inc. によってスピナーフラスコ中に遺伝子導入していない CHO-K1 細胞を使用して調製されたもので、2 ~ 15 日目にフラスコから使用済み培地を回収し(平均細胞生存率 約 90%)、プールして 0.2 µm フィルターでろ過しました。次に、スパイクを行った 2 サンプルを、Andrew+ ピペッティングロボット(Andrew Alliance™)を使用してプロテイン A 精製しました。簡単に説明すると、Pall の 0.2 µm Supor™ メンブレンフィルター付き 96 ウェル AcroPrep™ プレートに分注した Cytiva MabSelect™ レジン(約 15 µL)を使用してサンプル(各 120 µL)を捕捉しました。サンプルは、オービタルプレートシェーカー(1200 rpm)上で 20 分間結合させ、PBS で洗浄した後、30 µL の 100 mM グリシン(pH 3)で溶出し(静止保持時間 5 分間)、10 µL の 1.0 M TRIS(pH 7.5)で中和しました。回収率は、NTM スパイクサンプルで 94.4%(6.1 %RSD、n=8)、PBS スパイクサンプルで 90.8%(6.9 %RSD、n=8)でした。
SEC 分析法の有効性は、対照のトラスツズマブ-anns サンプルおよびプロテイン A 精製サンプルについて測定されたサイズバリアントの相対存在量によって実証されました(図 4)。SEC 分析法に関して、%RSD 値(n=3)は、HMWS1 では 0.9% RSD、LMWS1 では 2.4% RSD、微量レベルの LMWS2 では 7.9% RSD で、対照サンプル中のサイズバリアントの相対存在量が再現性よく測定されていることを示しています。2 セットのプロテイン A 精製サンプルについて得られた結果では、HMWS1(3.5% RSD)、LMWS1(2.8% RSD)、LMWS2(24.7% RSD)と最大変動がより大きくなりました(n=8)。これらの変動が大きいのは、プロテイン A 精製と SEC 分析法によって導入された変動の組み合わせに起因する可能性があります。
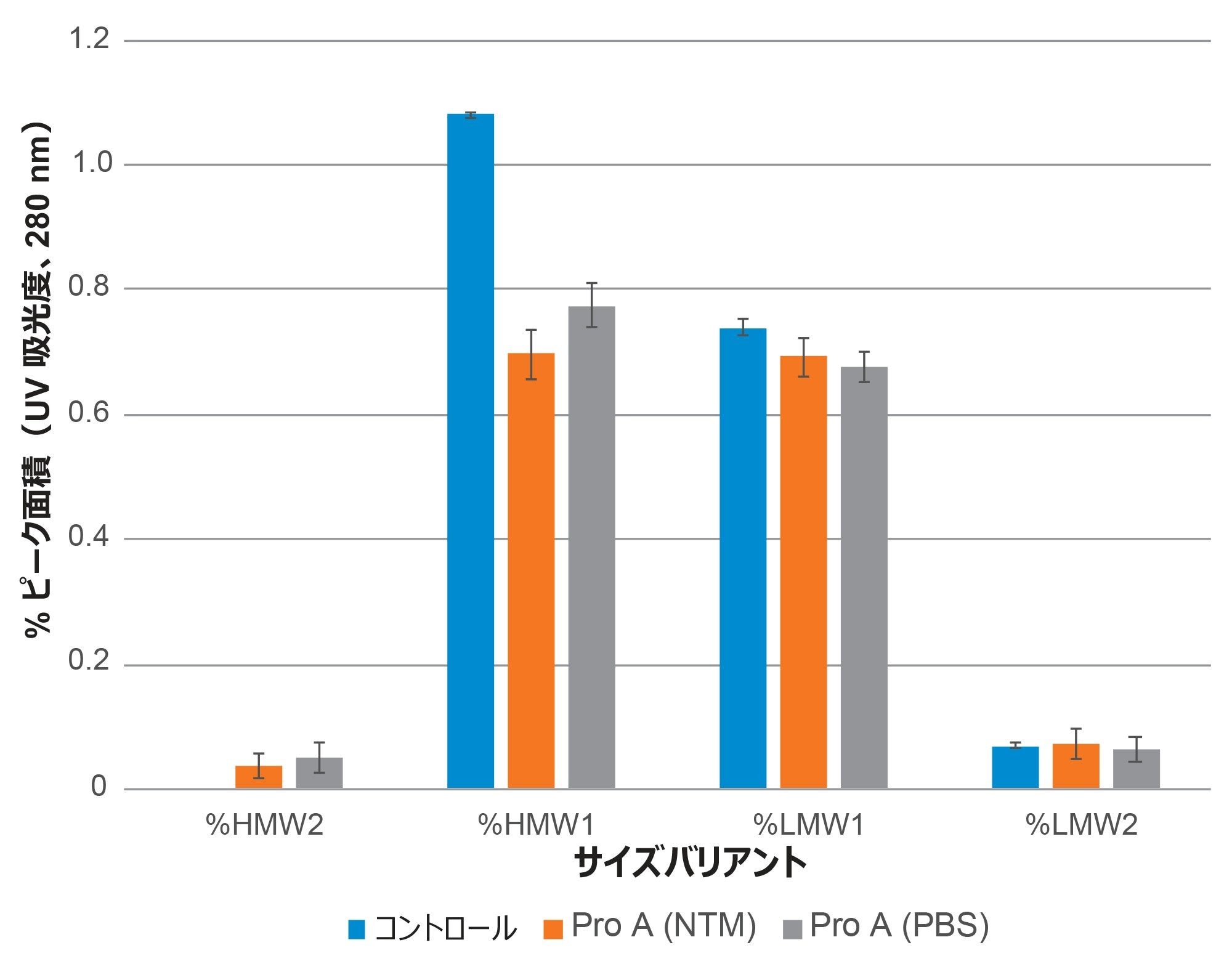 図 4. タンパク質 A 精製トラスツズマブ-anns の SEC-UV 定量結果の評価を示します。PBS(n=8)および遺伝子導入していない CHO 細胞でコンディショニングした培地(NTM、n=8)にスパイクしたトラスツズマブ-anns を、スパイクコントロール(n=3)と比較しています。クロマトグラフィーの条件は本文に記載しています。エラーバーは得られた値の範囲を表します。データは ACQUITY Premier BSM UPLC で収集しました。
図 4. タンパク質 A 精製トラスツズマブ-anns の SEC-UV 定量結果の評価を示します。PBS(n=8)および遺伝子導入していない CHO 細胞でコンディショニングした培地(NTM、n=8)にスパイクしたトラスツズマブ-anns を、スパイクコントロール(n=3)と比較しています。クロマトグラフィーの条件は本文に記載しています。エラーバーは得られた値の範囲を表します。データは ACQUITY Premier BSM UPLC で収集しました。
これ以上の調査はこの試験の範囲外でしたが、使用したプロテイン A ベースの mAb 精製手順は、トラスツズマブ-anns の HMWS の結果に多少のバイアスを生じさせています。微量(0.05% 未満)の HMW2 がアーティファクトとして生成し、HMW1 の回収が部分的になっています。HMW1 サイズバリアントの絶対回収率は、プロテイン A 精製プロセスで追加の HMW1 フォームが生じないと仮定すると、スパイクを行った NTM サンプルでは 68%、スパイクを行った PBS サンプルでは 59% と推定されました。プロテイン A アフィニティー精製を使用する場合の HMW mAb バリアントの定量的回収に関する課題は、より精度の高い LC ベースの方法を導入した場合でも生じることが以前に報告されています6。
全体として、これらの結果は、一般的なタンパク質の配合バッファー中でも中和したプロテイン A 溶離バッファー中でも、開発した SEC-UV 分析法が低サンプル濃度範囲(0.5 mg/mL 以上)の mAb サイズバリアントの測定に有効であることを示しています。最終的な分析法の分析時間は 4 分で、この間に、試験する mAb の HMW フォームを回収および分離でき、評価する複数の mAb の低分子量(LMW)サイズバリアントを分離することができます。この分離には ESI-MS 検出が使用できますが、AMA 濃度と流速が高いため、MS データの質が低下することに注意が必要です。
分析法の信頼性
分析法の信頼性の主要要素は、時間が経過しても保たれるカラム性能とカラム間再現性の 2 つです。3 か月以上にわたって 700 を超えるサンプルの分析に AMA 移動相を使用した場合のカラム寿命を計画外で評価しました(図 5)。この期間にわたって、カラムには、複数回の停止と開始や長期間使用しない時期があり、さまざまなプロテイン A 精製サンプル、部分精製サンプル、さらには粗細胞培養サンプルに曝されています。カラムの初期と後期のパフォーマンスを比較すると、初期の分離の多くが保持されているものの、全体的な保持にわずかなシフトが観察されました。多量体 HMW2 の存在量が少なくなっていることも分かります。ほぼ同時に同じトラスツズマブサンプルを使用し、このカラムと新しいカラムとで性能が同等であることを考慮すると、これは主にサンプルの安定性によるものと考えられます(図 6)。さらに、比較のために使用した新しいカラムにも、後で製造された粒子が充塡されていました。まとめると、これらの結果は、これらのカラムで高性能が維持され、信頼性の高いカラム間性能が得られることを示しています。
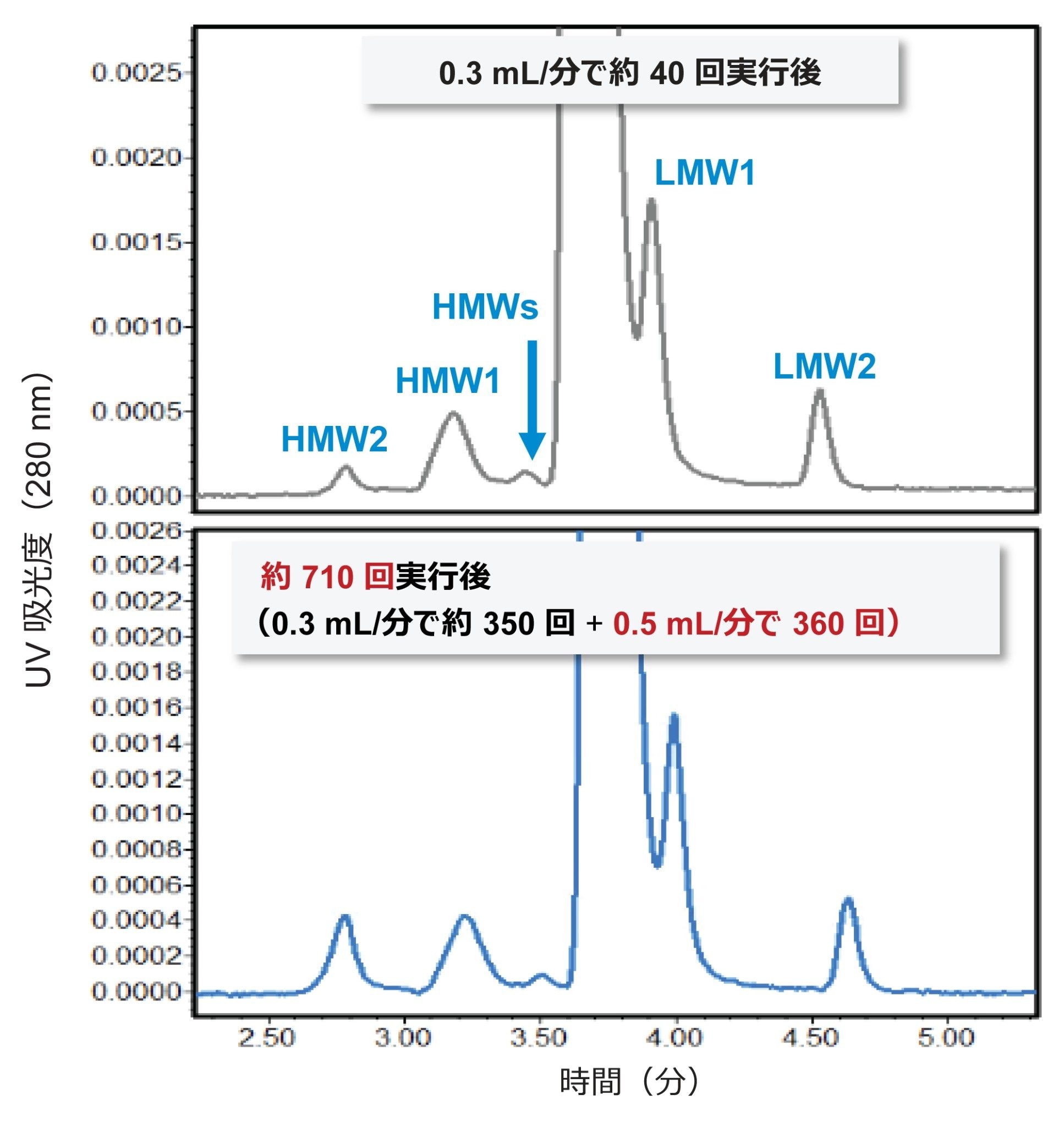 図 5. 200 mM 酢酸アンモニウム移動相を 0.3 ~ 0.5 mL/分の流速で使用する場合の、実使用での SEC カラムの寿命。トラスツズマブ(PBS 中 1 mg/mL)についての 3 か月間の 700 回を超える分析にわたるクロマトグラムの比較。クロマトグラフィーの条件および詳細なディスカッションは本文に記載しています。これらのデータは、ACQUITY Premier BSM UPLC で流速 0.3 mL/分で収集しました。
図 5. 200 mM 酢酸アンモニウム移動相を 0.3 ~ 0.5 mL/分の流速で使用する場合の、実使用での SEC カラムの寿命。トラスツズマブ(PBS 中 1 mg/mL)についての 3 か月間の 700 回を超える分析にわたるクロマトグラムの比較。クロマトグラフィーの条件および詳細なディスカッションは本文に記載しています。これらのデータは、ACQUITY Premier BSM UPLC で流速 0.3 mL/分で収集しました。
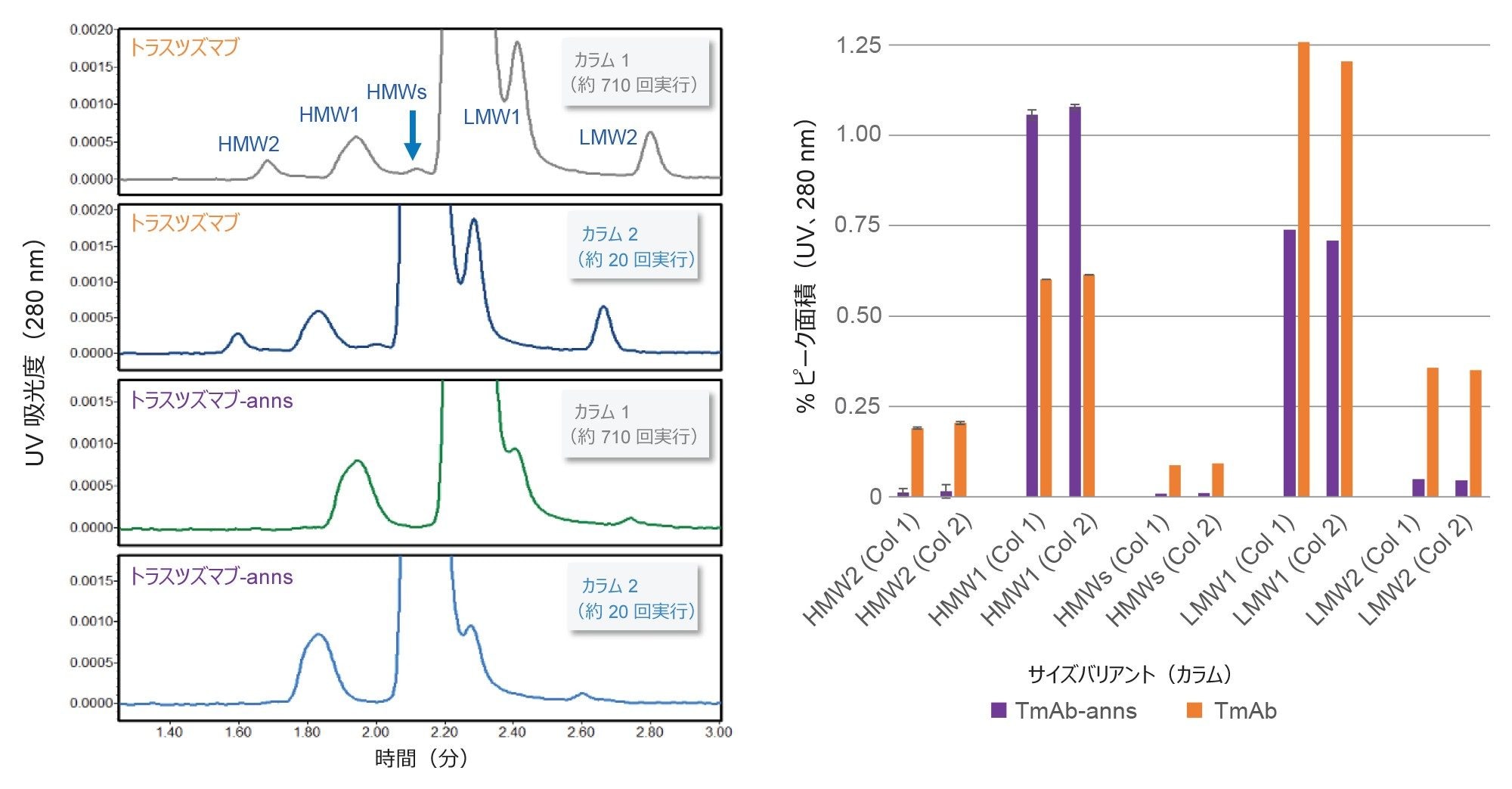 図 6. カラム間再現性が、数か月隔てて製造された異なるロットの SEC 粒子を充塡した 2 本のカラムで実証されました。評価したサンプルは、1 mg/mL(PBS)のトラスツズマブ-anns およびトラスツズマブです。200 mM 酢酸アンモニウム移動相を流速 0.5 mL/分で使用しました。カラム 1 は 3 か月間で 700 回以上の分析に使用され、カラム 2 のデータは 20 回の分析の後に生成されました。クロマトグラフィーの条件および詳細なディスカッションは本文に記載しています。エラーバーは得られた値の範囲を表します(n=2)。データは ACQUITY Premier BSM UPLC で収集しました。
図 6. カラム間再現性が、数か月隔てて製造された異なるロットの SEC 粒子を充塡した 2 本のカラムで実証されました。評価したサンプルは、1 mg/mL(PBS)のトラスツズマブ-anns およびトラスツズマブです。200 mM 酢酸アンモニウム移動相を流速 0.5 mL/分で使用しました。カラム 1 は 3 か月間で 700 回以上の分析に使用され、カラム 2 のデータは 20 回の分析の後に生成されました。クロマトグラフィーの条件および詳細なディスカッションは本文に記載しています。エラーバーは得られた値の範囲を表します(n=2)。データは ACQUITY Premier BSM UPLC で収集しました。
結論
迅速非変性サイズ排除クロマトグラフィー(SEC)分析法は、バイオ医薬品の細胞培養および精製プロセスの開発、ならびに原薬および薬物製剤の開発の支援に有用です。さらに、酢酸アンモニウム移動相を使用する迅速 SEC-UV 分析法も、非揮発性の塩およびバッファー(NaCl、リン酸など)で構成される従来の SEC の移動相を使用するよりも、LC-MS 分析専用の LC システムまたはカラムにより効率的に導入できます。
具体的には、このアプリケーションでは、流速 0.50 mL/分の 200 mM 酢酸アンモニウム移動相および UV 吸光度検出(280 nm)を使用する ACQUITY Premier Protein SEC(250 Å、1.7 µm、4.6 × 150 mm)カラムでの、モノクローナル抗体のサイズバリアント(凝集およびフラグメンテーション)分析に用いるハイスループット(分析時間 4 分)SEC-UV 分析法を最適化することに成功しました。信頼性の高い分析法の性能について、BioAccord LC-MS(ESI-Tof)システムに加えてスタンドアローンの ACQUITY Premier UPLC システムも評価し、両方とも拡散容量の低いシステムでした。
さらに、3 か月間にわたる 700 回以上の分析でのカラム寿命の性能、およびカラム間の再現性も実証されました。この評価では、移動相は 0.1 µm フィルターで滅菌ろ過し、サンプルは個別に、または使用したプロテイン A 精製手順の一部として、0.2 µm フィルターでろ過しました。これにより、カラムが汚れる可能性が大きく低減します。ただし、顕微鏡で見える微粒子やより大きい微粒子が分析する開発サンプルに大量に含まれている場合は、ガードカラム(MaxPeak™ Premier Protein SEC Guard、250 Å、製品番号:186009969)や遠心分離などのサンプル前処理の使用が推奨されます。
参考文献
- Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Nari LO, Topp EM.“Immunogenicity of Therapeutic Protein Aggregates”.J Pharm Sci.2016 Feb;105(2):417–430.
- Stephan M. Koza, Hua Yang, and Ying Qing Yu, “Expanding Size-Exclusion Chromatography Platform Method Versatility for Monoclonal Antibody Analysis Using Waters XBridge Premier Protein SEC Columns”, Waters Application Note, 720007500, January 2022.
- Stephan M. Koza and Ying Qing Yu, “Rapid Size Variant Analysis of Monoclonal Antibodies Using UPLC™ and HPLC Compatible MaxPeak™ Premier Protein SEC Columns”, Waters Application note, 720007584, March 2022.
- Pinhal, S., Ropers, D., Geiselmann, J. and De Jong, H., 2019.Acetate metabolism and the inhibition of bacterial growth by acetate.Journal of bacteriology, 201(13), pp.e00147–19.
- Stephan M. Koza, Corey Reed, and Weibin Chen, “Impact of LC System Dispersion on the Size-Exclusion Chromatography Analysis of Monoclonal IgG Antibody Aggregates and Fragments: Selecting the Optimal Column Configuration for Your Method”, Waters Application note, 720006336, June 2019.
- Dunn ZD, Desai J, Leme GM, Stoll DR, Richardson DD.Rapid two-dimensional Protein-A size exclusion chromatography of monoclonal antibodies for titer and aggregation measurements from harvested cell culture fluid samples.MAbs.2020;12(1):1702263.
720007852JA、2022 年 2 月