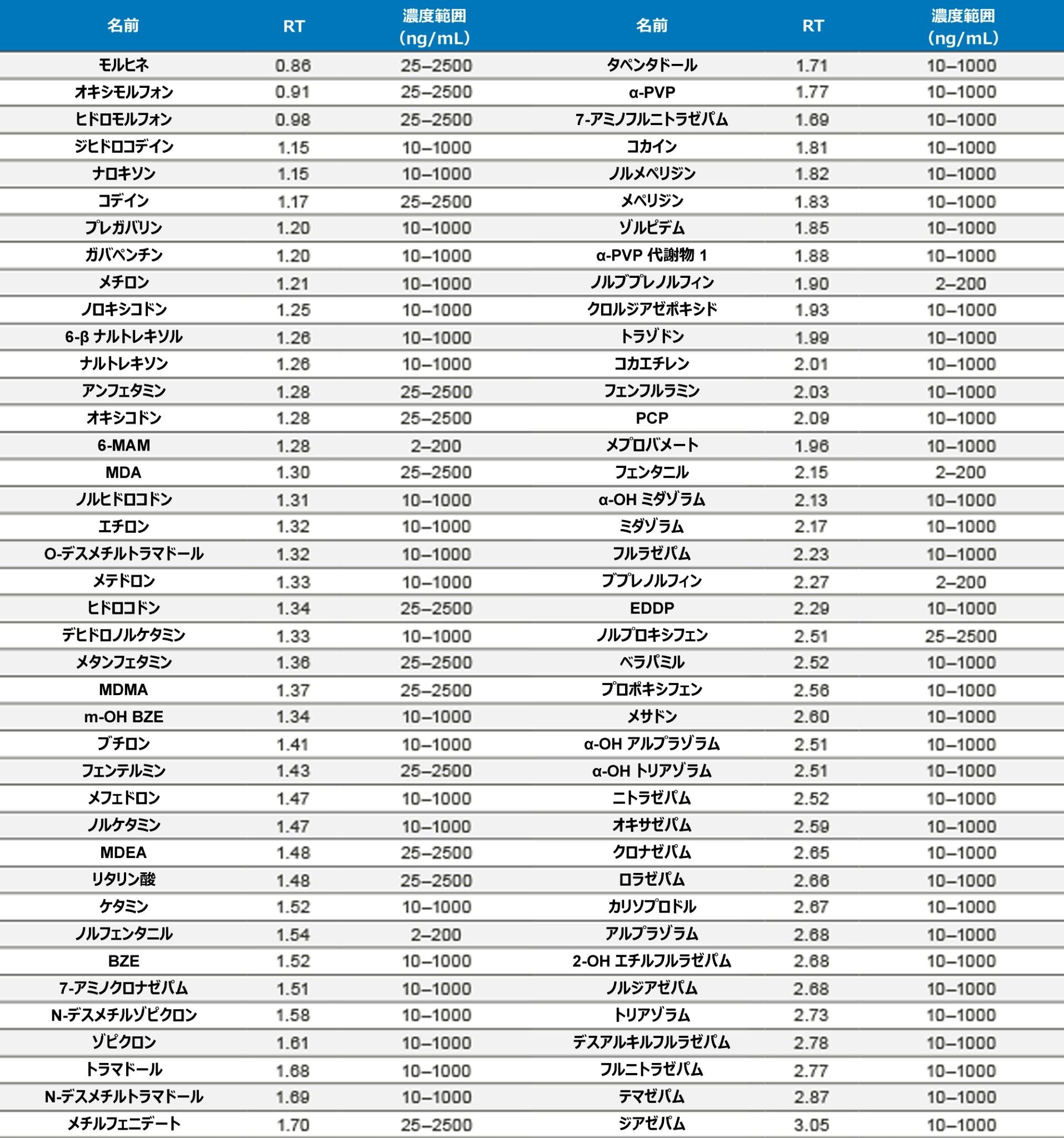
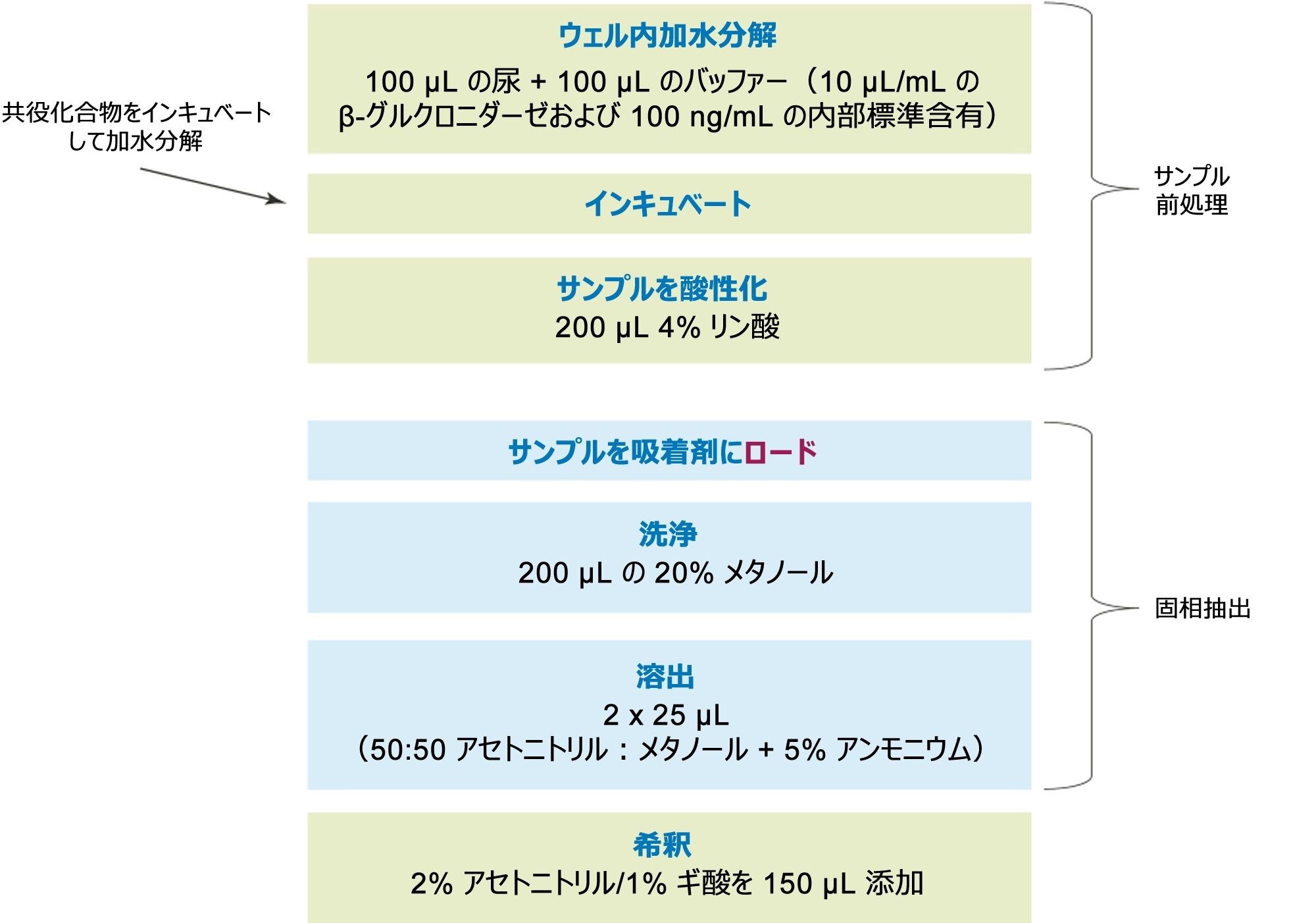
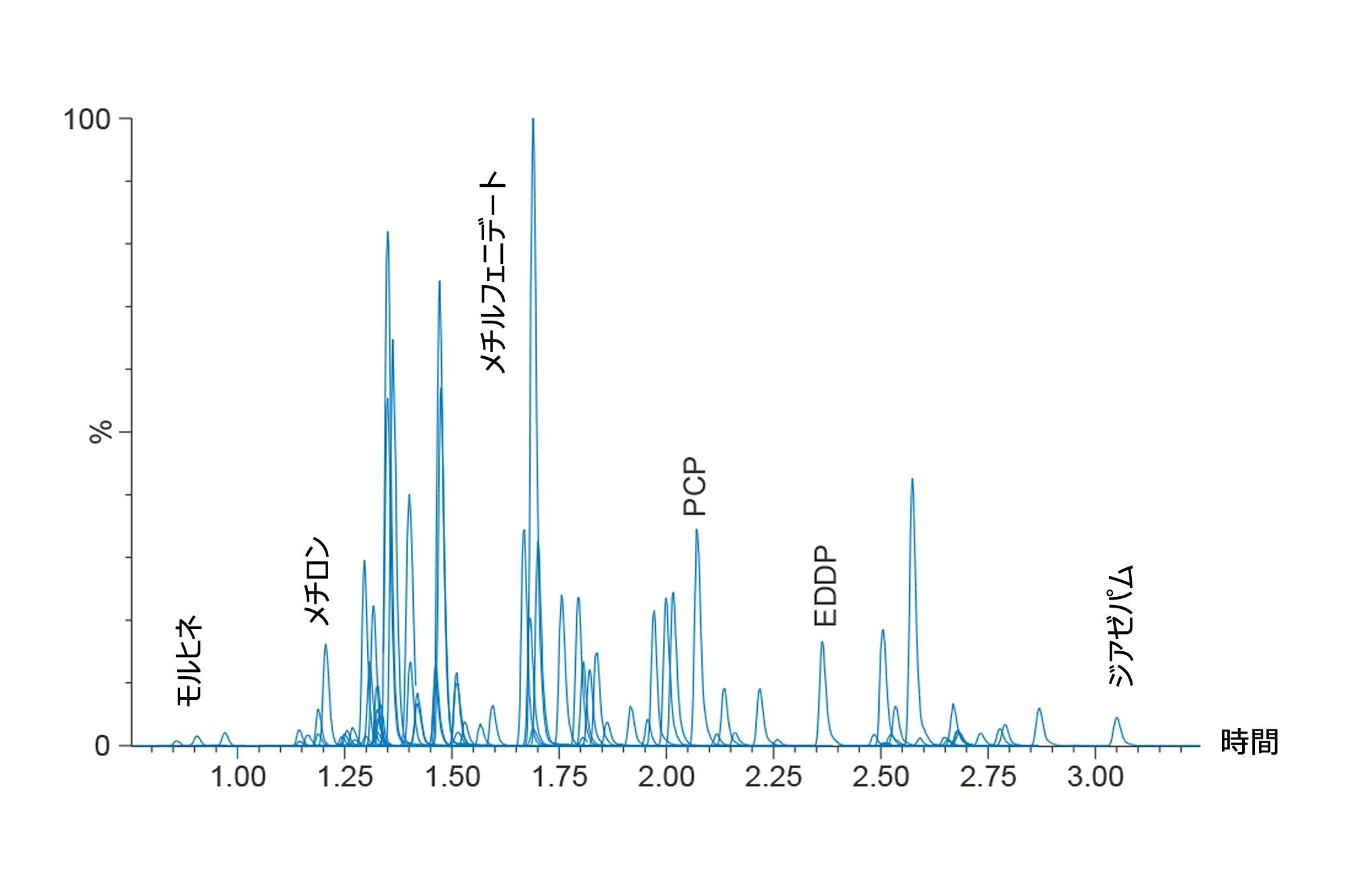
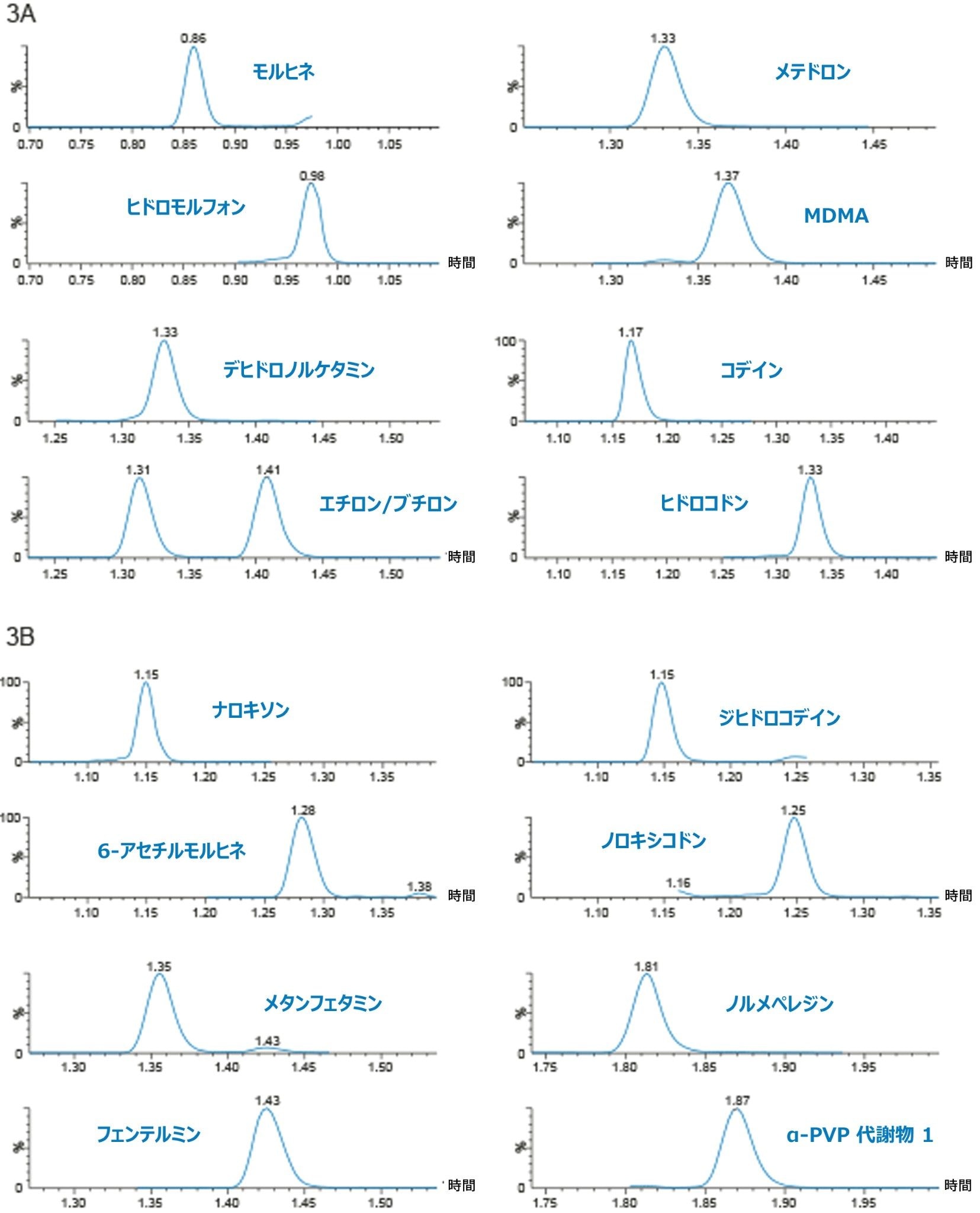
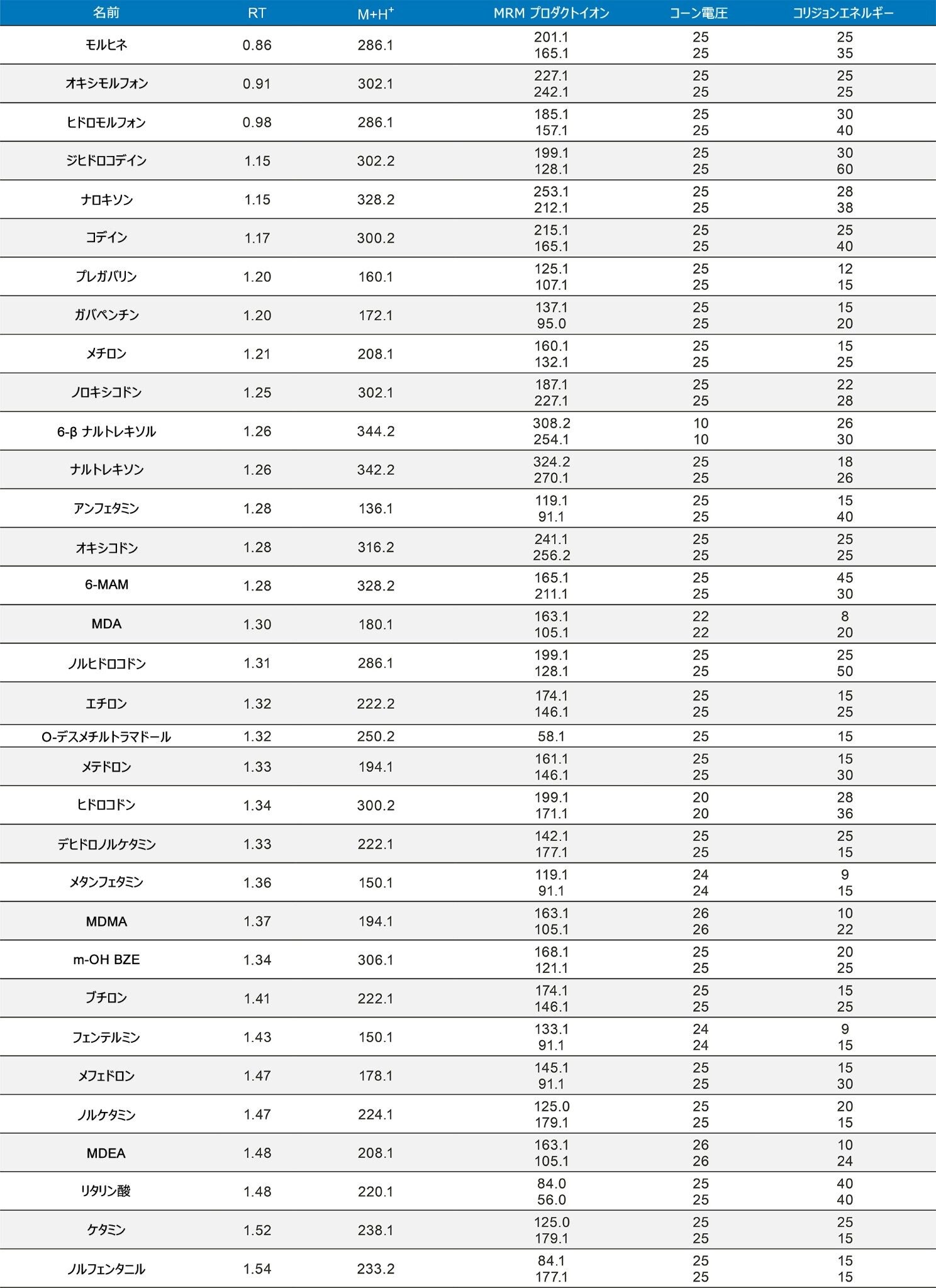
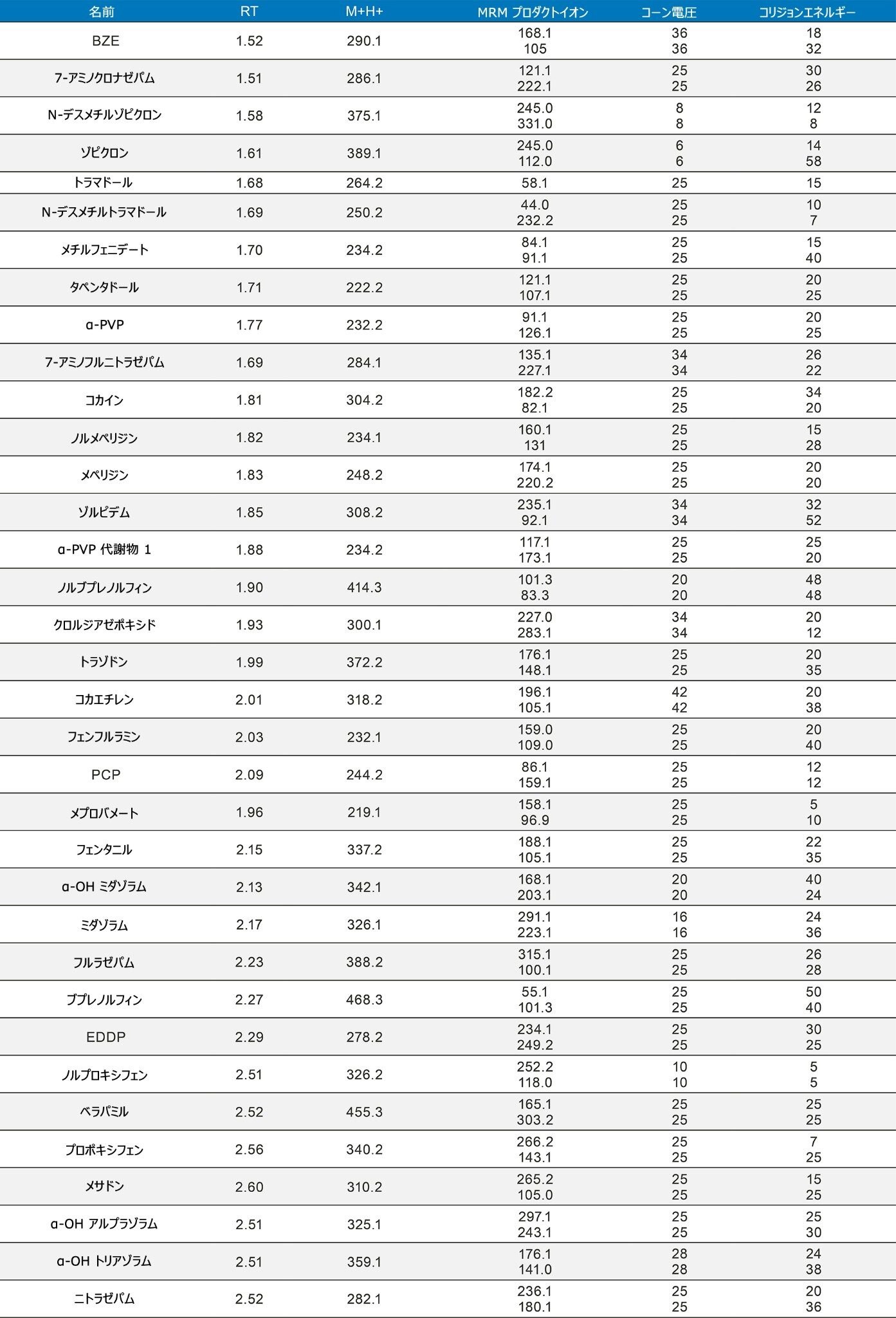
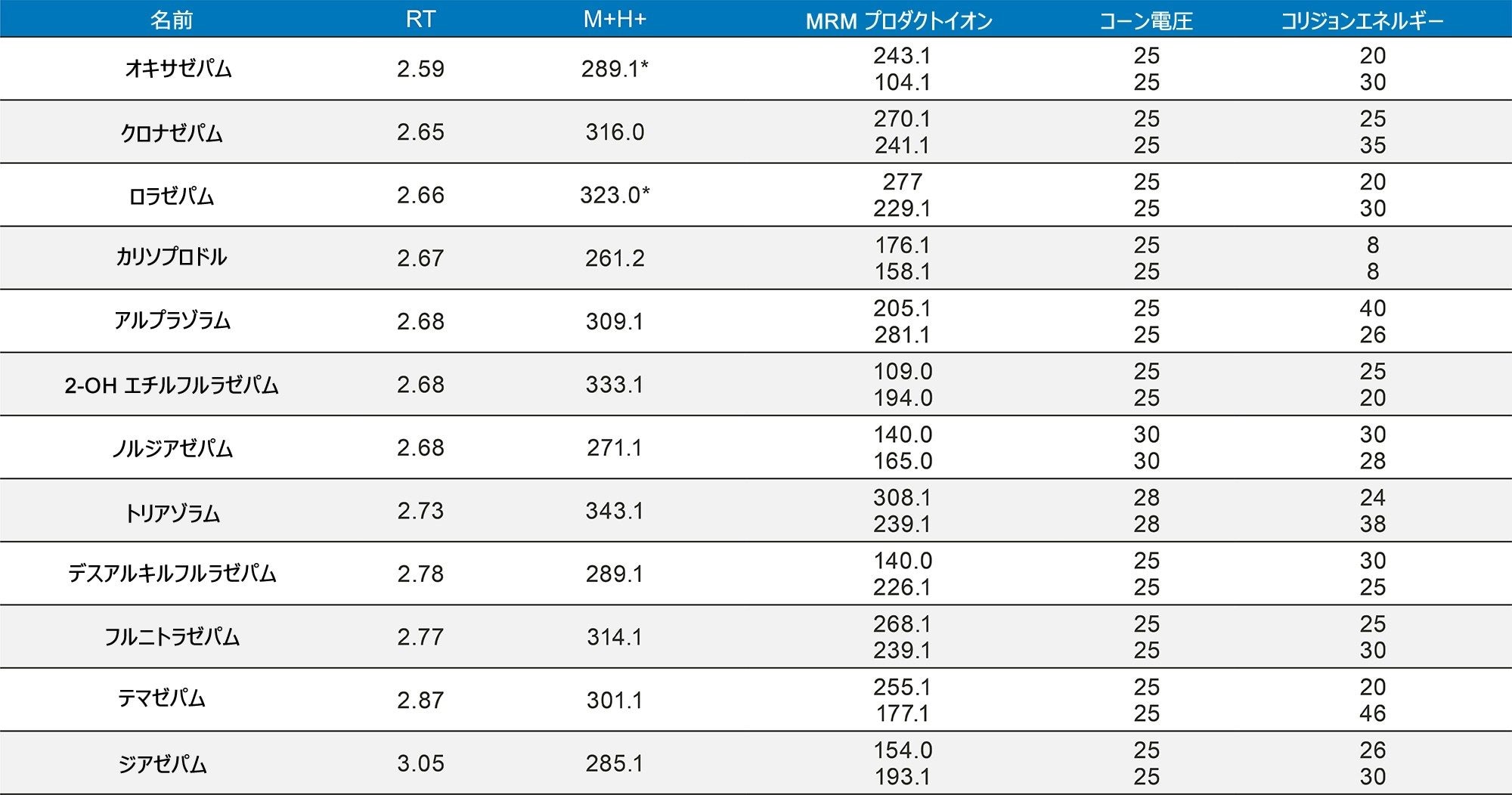
定量分析
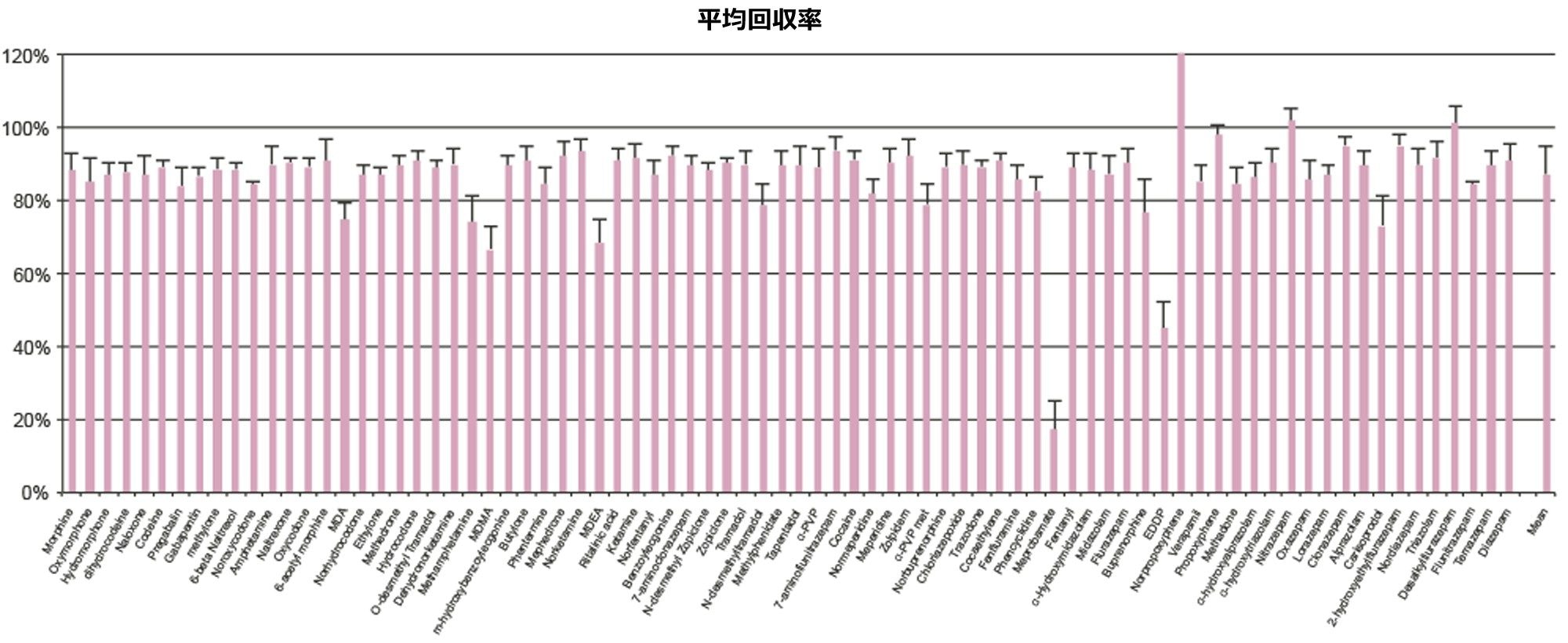
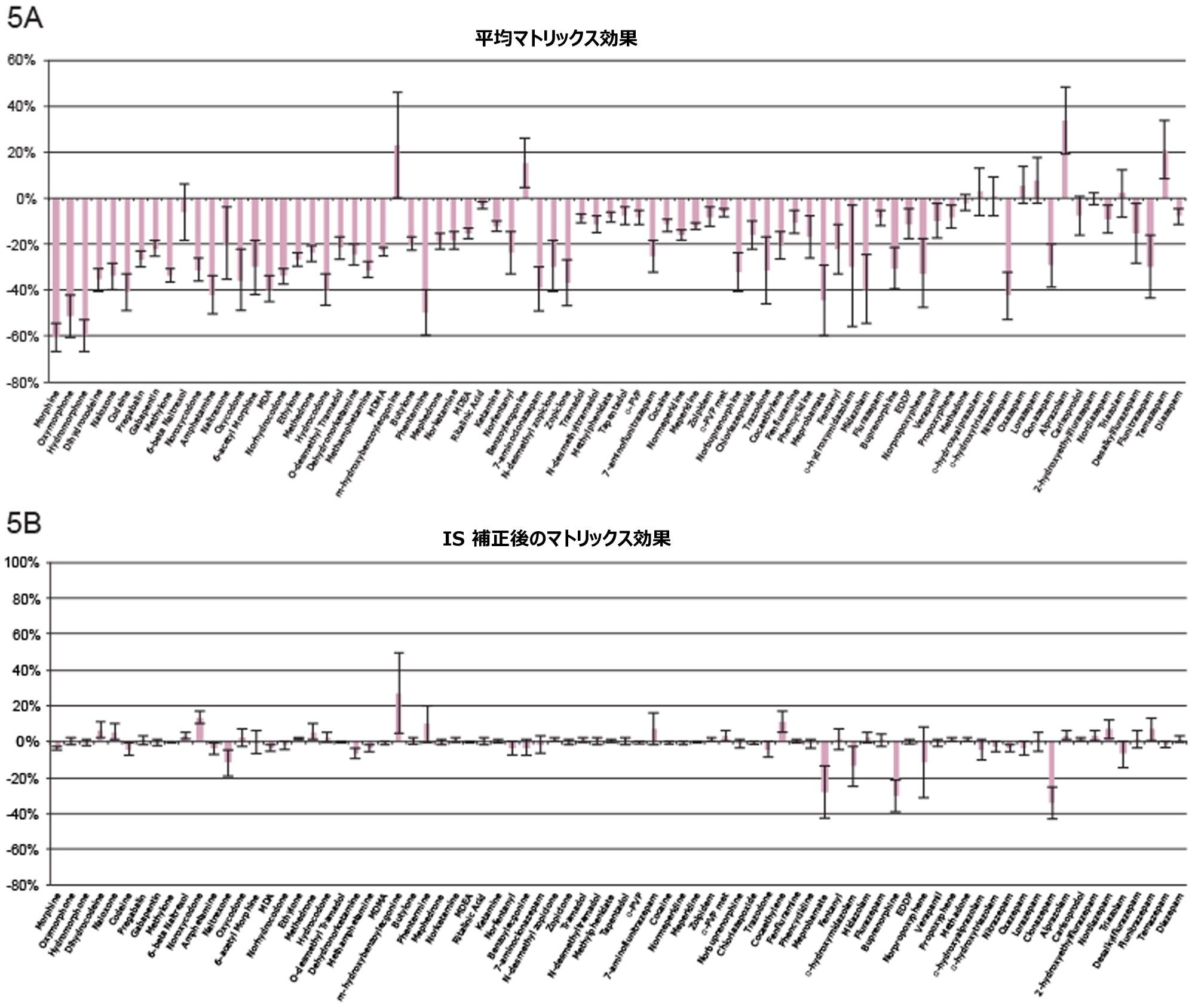
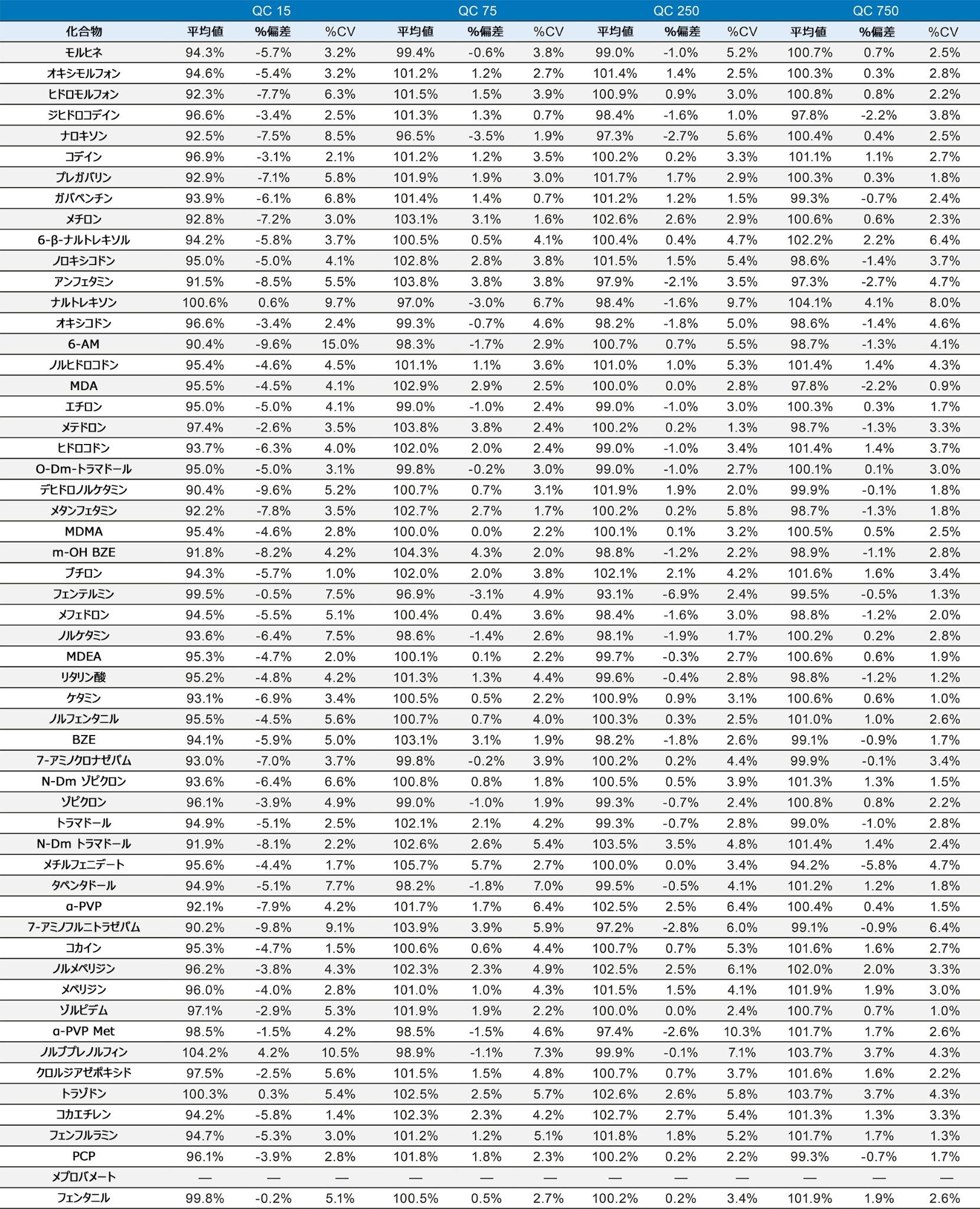
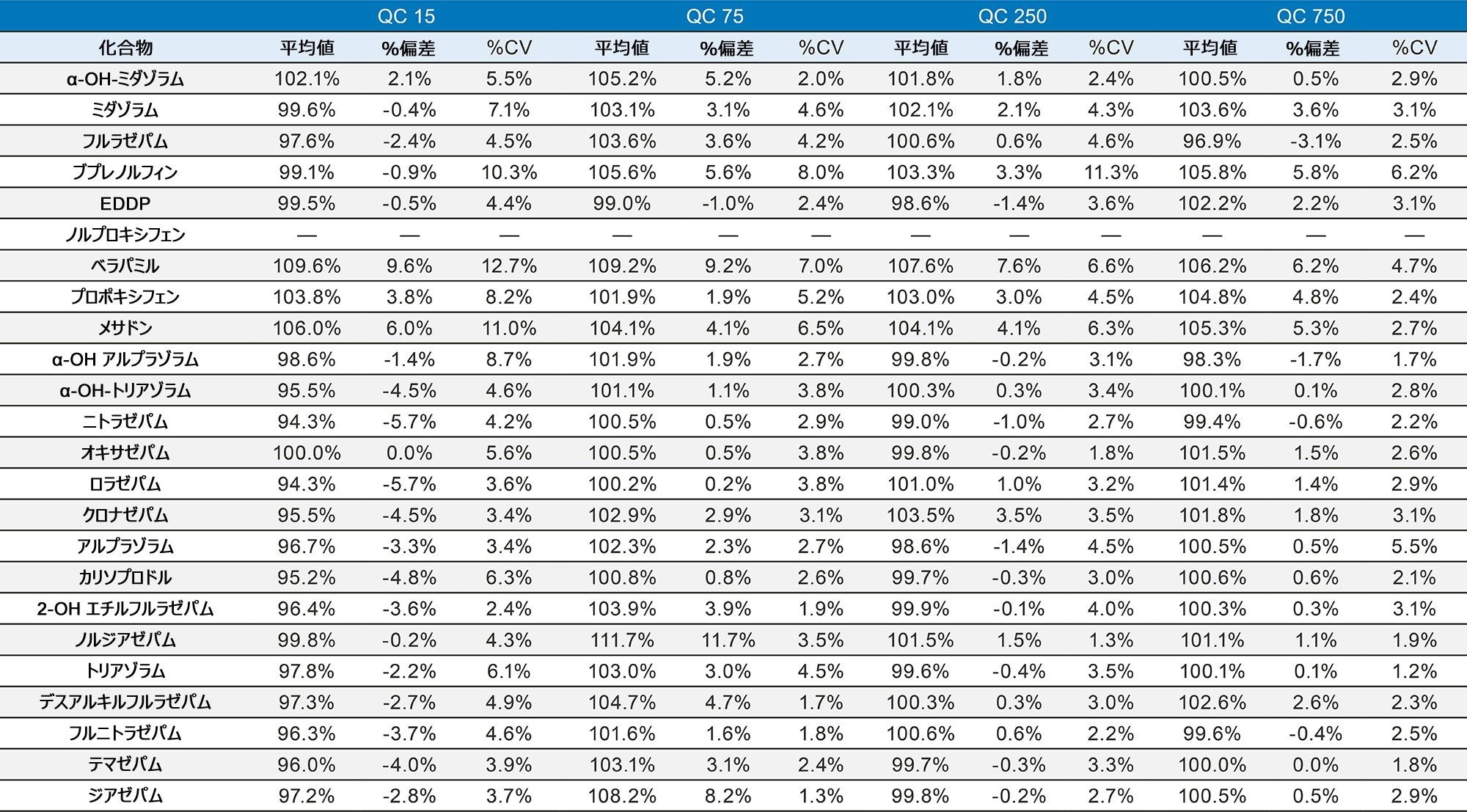
表 1 に示す濃度範囲にわたって、7 点検量線を抽出しました。キャリブレーション範囲を調整して、さまざまな化合物の予想濃度が反映されるようにしました。品質管理サンプルは、キャリブレーション試薬の範囲にまたがる 4 つの濃度で調製しました。最低試料では最低キャリブレーション試薬の 1.5 倍、最高試料では最高キャリブレーション試薬の 75% になるように調製しました。ほとんどの化合物での QC レベルは 15、75、250、および 750 ng/mL でした。低濃度の化合物での QC レベルは 3、15、50、150 ng/mL で、高濃度範囲の分析種の QC レベルは 37.5、187.5、625、1875 ng/mL でした。定量メソッドのバリデーションでは、5 日間にわたって完全な検量線および QC サンプルの抽出を行いました。検量線を 2 回、QC サンプルを 6 回抽出し、調製を毎日行いました。個々のキャリブレーション試薬と QC サンプルのコントロール限界は、ターゲット値の ±15% でした。ただし、最低点についてのみ、20% 以内であることが要求されました。QC サンプルの精度限界は、最低 QC ポイントについては 20%、その他のポイントについては 15% でした。メプロバメートとノルプロポキシフェンは定性的評価のみを行い、これらのコントロールの対象にはしませんでした。5 回の独立した抽出および分析のサマリーでは、これらの基準がすべて満たされていました(付録 2 参照)。大部分の化合物がターゲット値の 10% 以内であり、%CV は 10% 未満でした。バッチ内結果では、すべての化合物が正確性基準を満たしており、15% を超える精度結果が得られた唯一の化合物は、アンフェタミンの高 QC(18%)でした。
すべての検量線は FDA のバイオアナリシス分析法のバリデーション要件に準拠しています3。この要件では、すべてのキャリブレーション試薬は、ターゲット値の 15% 以内に収まることが求められます(ただし、最低点はターゲット値の 20% 以内)。キャリブレーション試薬の 75% でこの基準が満たされています。すべての化合物がこれらの基準に適合しており、すべての曲線の R2 値は 0.99 以上を示していました。
定量限界は、シグナルが抽出マトリックスブランクの 5 倍であり、シグナル対ノイズ比が 10 を超え、バイアスと %CV が両方とも 20% 未満であるポイントとして定義されました。この点を評価するために、いずれかのバリデーションバッチで最低のキャリブレーション試薬を 6 回繰り返し抽出しました。すべての化合物がこれらの基準に適合していました。
装置での安定性も評価しました。単一バッチの抽出と分析は、8 日間にわたって 5 回行われました。4 日間にわたって、すべての化合物が上記の定量バリデーション基準に適合していました。
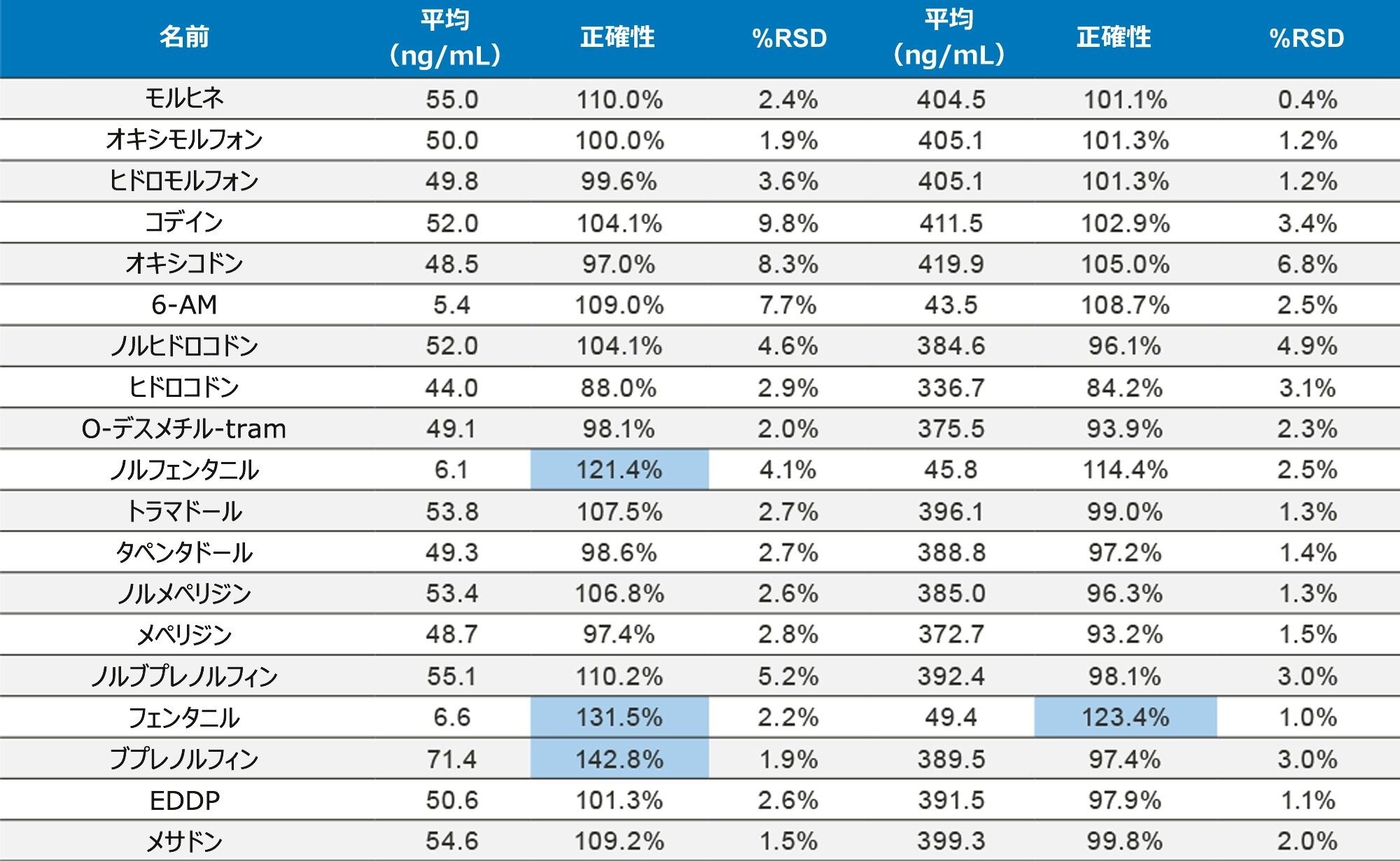
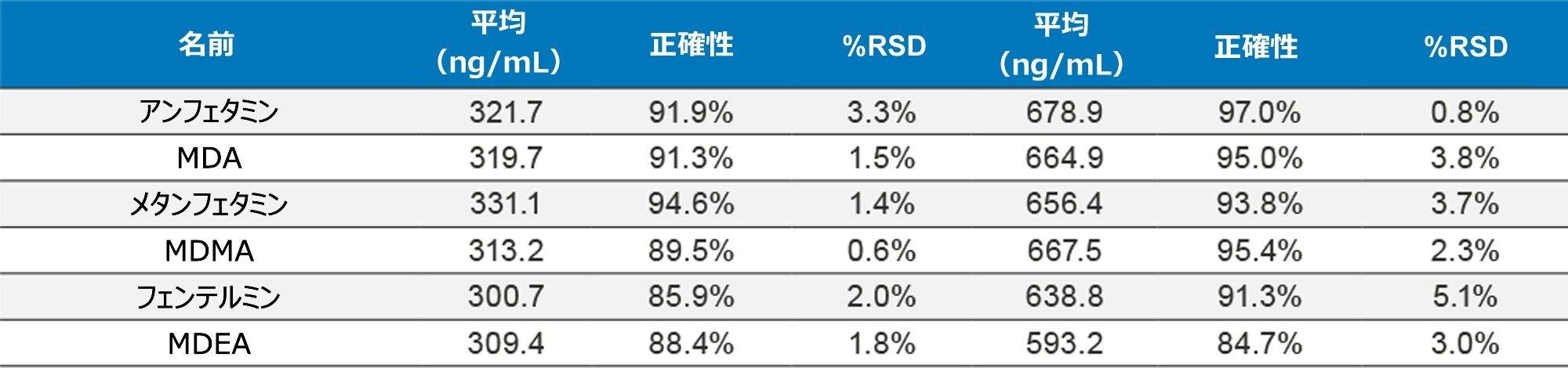
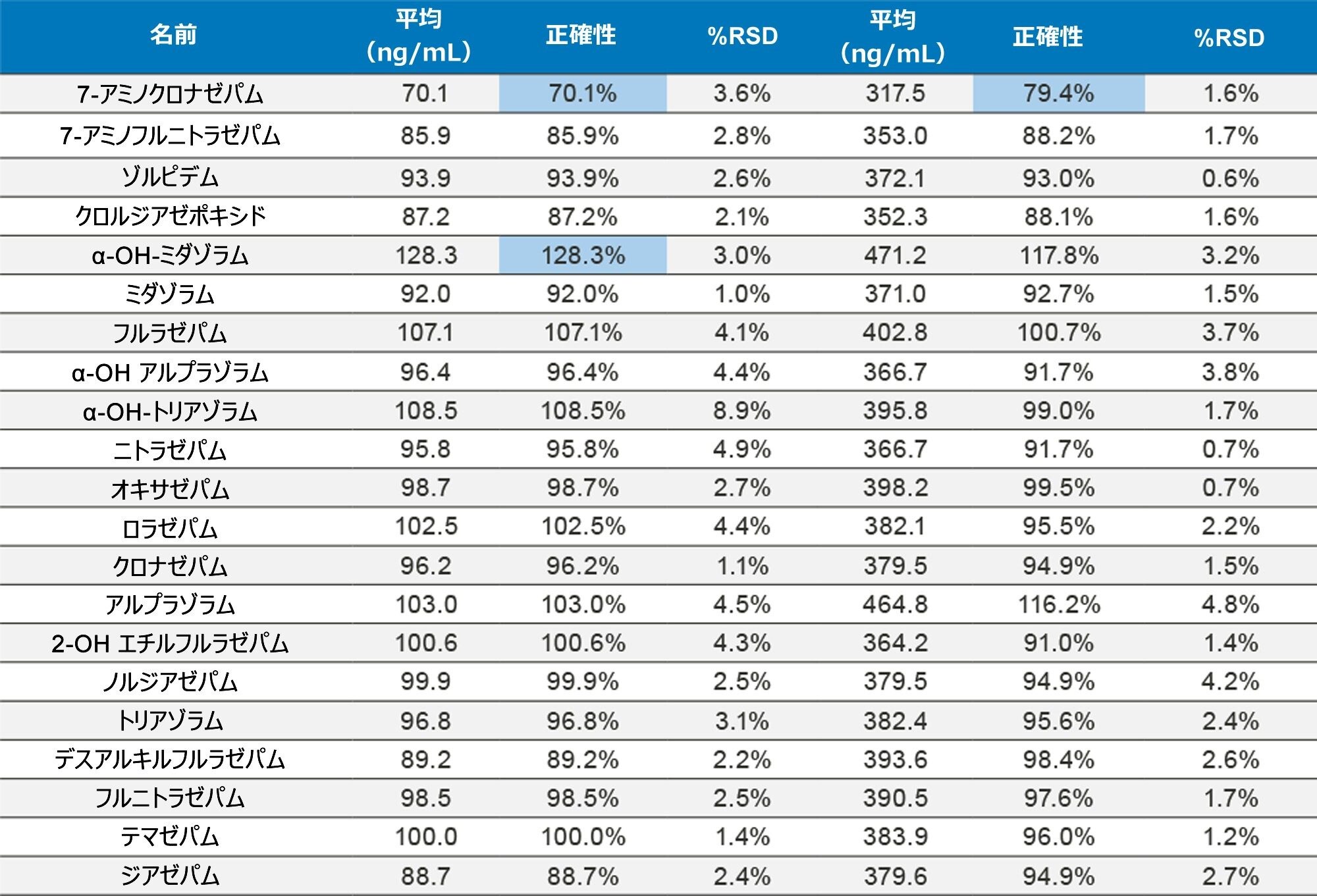
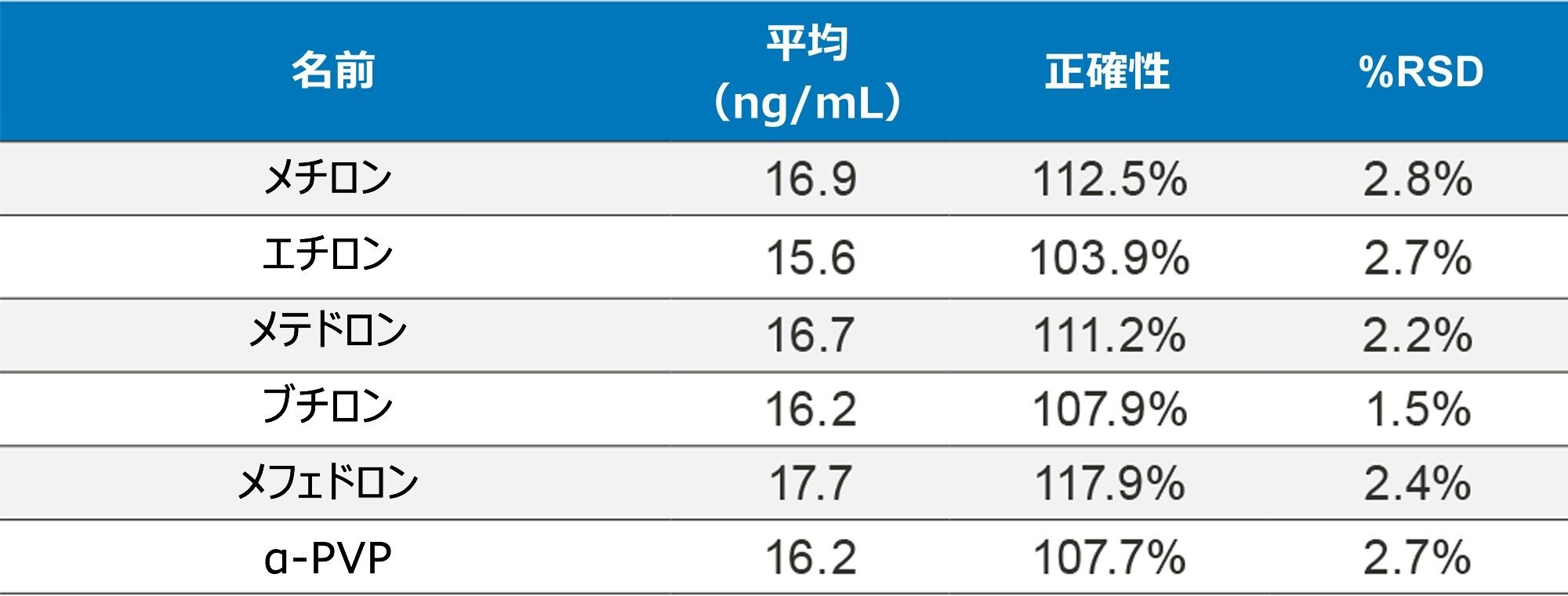
正確性を評価するために、UTAK Laboratories からの外部品質管理サンプルを評価しました。これらの結果を表 2A ~ 2D にまとめています。外部品質管理サンプルを使用して評価した分析種には、オピオイド、ベンゾジアゼピン、覚せい剤、合成カチノンが含まれていました。これらの結果からは、91/98(93%)の結果がターゲット値の 20% 以内であることを示しています。フェンタニル、ノルフェンタニル、ブプレノルフィンなどの化合物は、低容量(20 µL のストック溶液)を使用してスパイクされているため、これらの分析種での大きな偏差は、マスターストック混合液の調製におけるわずかなミスの結果である可能性があります。さらに、7-アミノクロナゼパムには、尿マトリックス中での安定性の問題が生じる可能性があり、これが低バイアスの原因となる可能性があります。すべての結果において %RSD 値は 10% 未満でした。