Qualitative and Quantitative Performance of Cyclic IMS in Nanoscale Proteomic Experiments
This is an Application Brief and does not contain a detailed Experimental section.
For research use only. Not for use in diagnostic procedures.
Abstract
The performance characteristics of Cyclic IMS in qualitative discovery and quantitative proteomic experiments are described. All the experiments consisted of a single pass around the cyclic mobility device, with the electrospray source coupled to a nanoscale LC Inlet.
The results shown for discovery experiments are based on the analysis of standard cell lysate samples, including E. Coli and Human K562, while for dynamic range/quantitation experiments we used processed data from qualitative identifications plus standard peptide mixtures with and without complex background matrix. The results highlight performance improvements when compared to previous iterations of QTof Mass Spectrometers (MS).
Benefits
- Ion mobility resolution
- Mass resolution increase
- Enhanced qualitative/quantitative performance over previous QTof iterations
Introduction
Quadrupole Time-of-Flight (QTof) Mass Spectrometers are a well-established tool for both discovery and quantitative proteomic experiments. These instruments display sensitivity, speed, and high mass resolution, which are important characteristics required for the successful analysis of these challenging types of sample(s). SELECT SERIES Cyclic IMS is a recent addition to the QTof family of instruments and provides enhanced sensitivity, ion mobility resolution, and mass resolution when compared to previous iterations of QTof platforms.1 In addition, the Cyclic IMS exhibits extended dynamic range due to different detector characteristics, with the instrument utilizing a dual gain detection system.
In a classical proteomic experiment, sample amounts tend to be limited and require the coupling of the MS source to a nanoscale chromatography inlet. Doing so provides a powerful combination, enabling the generation of high quality, information-rich datasets which can then be processed to provide metrics for both qualitative and quantitative performance. We have assessed the Cyclic IMS in the analyses of proteomics, incorporating standard tryptic digest samples, varying in complexity from relatively simple peptide mixtures to more complex human cell lines.
Experimental
Sample Description
Qualitative: Waters MPDS E. Coli (p/n 186003196) and Human Cell Lysate K562 (Promega, V6951) digests were reconstituted to a concentration of 100 ng/µL in aqueous 0.1% formic acid. Samples were loaded onto the column from 10 to 100 ng, with multiple injections performed at each loading level.
Quantitative: 6x5 isotopologue (Promega, V7495) mixture was prepared in E. Coli background matrix.
LC Conditions
|
LC system: |
ACQUITY UPLC M-Class |
|
Trapping column: |
Symmetry, 180 micron x 20 mm (p/n 186008821) |
|
Analytical column: |
HSS T3, 75 micron x 250 mm (p/n 186008818) |
|
Column temp.: |
35 °C |
|
Sample temp.: |
10 °C |
|
Flow rate: |
300 nL/min |
|
Mobile phase A: |
Aqueous 0.1% formic acid |
|
Mobile phase B: |
Acetonitrile + 0.1% formic acid |
|
Trapping conditions: |
Two minutes at 5 µL/min, 99% solvent A |
|
Gradient: |
5% to 35% mobile phase B over various gradient lengths, ranging from 90-minutes up to 240-minutes |
MS Conditions
|
MS system: |
SELECT SERIES Cyclic IMS |
|
Ionization mode: |
ESI+ |
|
Mass resolution: |
50000 FWHM |
|
Ion mobility resolution: |
Single pass, 65 FWHM |
|
Acquisition mode: |
HDMSE – Data Independent Acquisition |
|
Acquisition mass range: |
50–2000 Da |
|
Integration time: |
0.5 seconds |
|
Reference material: |
Glu Fibrinopeptide B sampled every 120 seconds |
|
Capillary voltage: |
3.2 kV |
|
Transfer CE, function 2: |
19–45 V (linear ramp) |
|
Cone voltage: |
30 V |
Data Management
|
MS software: |
MassLynx |
|
Data processing: |
ProteinLynx Global Server, Progenesis QI for proteomics |
|
Databases: |
Uniprot E. Coli and Human - reviewed sequences only |
|
False discovery rate: |
1% and 4% |
Results and Discussion
During HDMSE, the acquisition alternatively produces low energy and elevated energy chromatograms, plus a lock mass reference channel. Figure 1 shows a representative example of the low and elevated energy chromatograms from the complex K562 human cell line digest.
The elevated energy fragmentation occurs in the transfer collision cell, which is located after the ion mobility separation, hence precursors and fragments exhibit the same drift time information. Therefore, during data processing, the fragment ions from function 2 (elevated) are aligned with precursor information (function 1), based upon both retention and drift time information. The aligned data is then constructed into a binary file which can then be readily database searched.
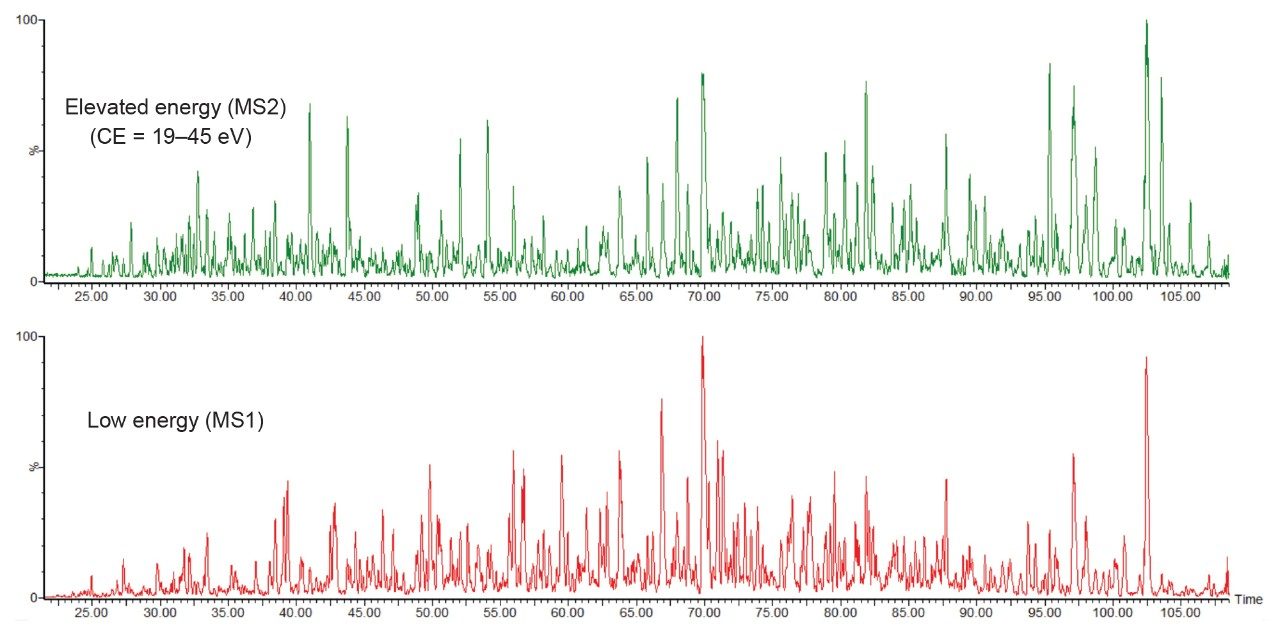 Figure 1. Low and elevated energy chromatograms representing the complex K562 human cell line digest. Sample loading on-column was 75 ng and separated over a 90-minute gradient.
Figure 1. Low and elevated energy chromatograms representing the complex K562 human cell line digest. Sample loading on-column was 75 ng and separated over a 90-minute gradient.
Figure 2 shows results corresponding to the Human K562 lysate as a result of database searching. Protein identifications representing protein groups, whereby homology is collapsed and the total number of proteins, whereby homology is included, are shown. The corresponding peptide identifications follow a similar pattern to that observed for the number of identified proteins, which maximizes at approximately 60000 peptides (data not shown). The identifications provided in Figure 2, represent database searches consisting of an FDR of 1% and 4%, based on sample loadings from 25 to 100 ng. Based on a 90-minute gradient, the data suggests that a loading of 50–75 ng is optimum. A similar pattern of protein and peptide identifications reaching a plateau is also observed for E. Coli, with an optimum of 50 ng on column, which corresponded to 1250 protein/20000 peptide identifications. By increasing gradient length, peptide ions become more separated in retention time and hence data processing algorithms are more easily able to distinguish between ions. The K562 sample was injected using different gradient lengths and Figure 3 shows the protein identifications returned at 1% FDR, increasing from 3700 (90-minute separation) to 4500 (240-minute separation). Major contributing factors to the identification rates obtained through the processing are the excellent mass accuracy of the system and dynamic range of the species identified. Typically, for both K562 (Figure 4) and E. Coli analyses, over 90% of peptide ions are mass measured to within +/-5 ppm, with identifications spanning 5 orders of dynamic range, Figure 5.
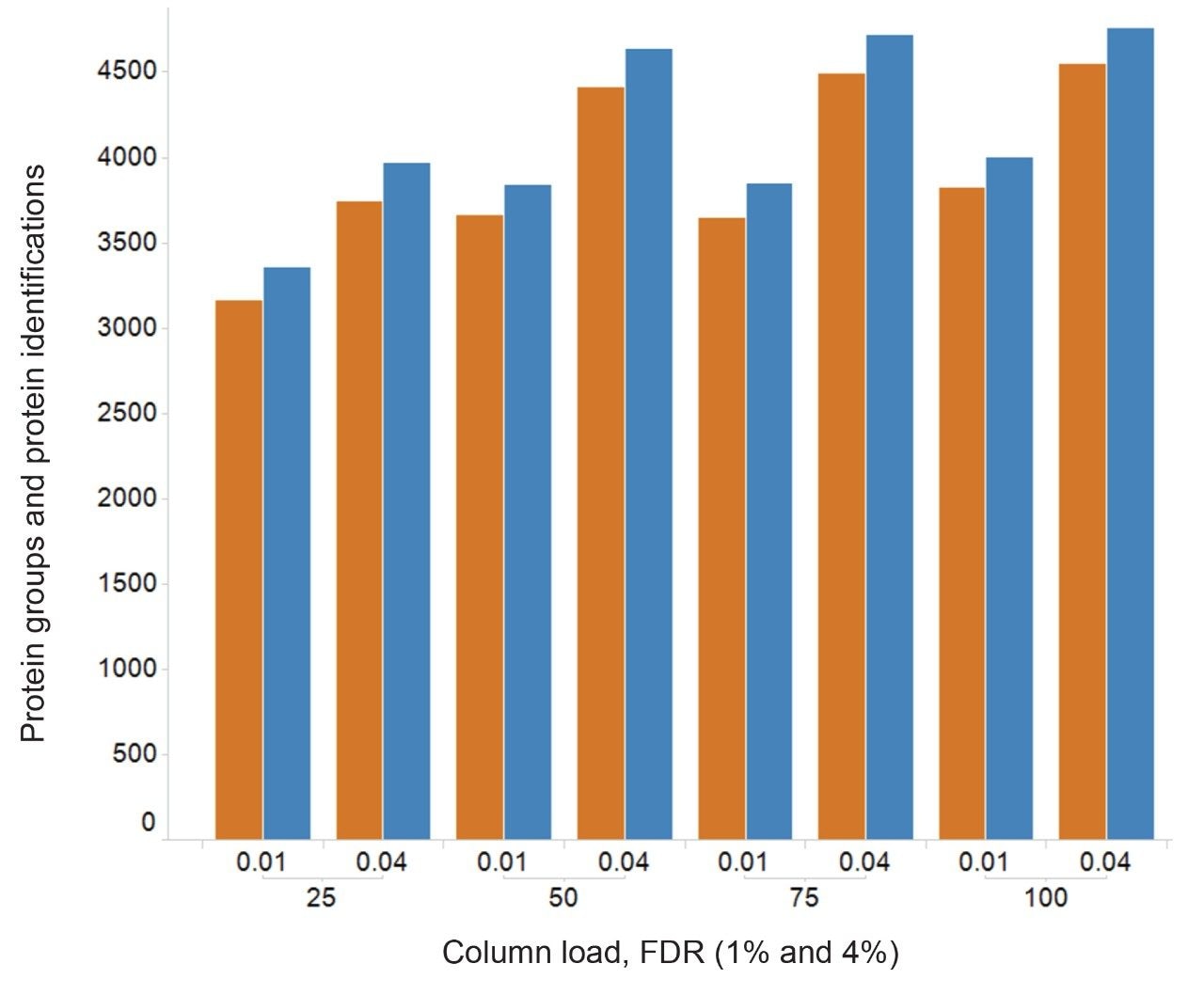 Figure 2. Protein identifications at an FDR of 1 and 4% for K562 loads from 25 to 100 ng, using 90-minute gradients.
Figure 2. Protein identifications at an FDR of 1 and 4% for K562 loads from 25 to 100 ng, using 90-minute gradients.
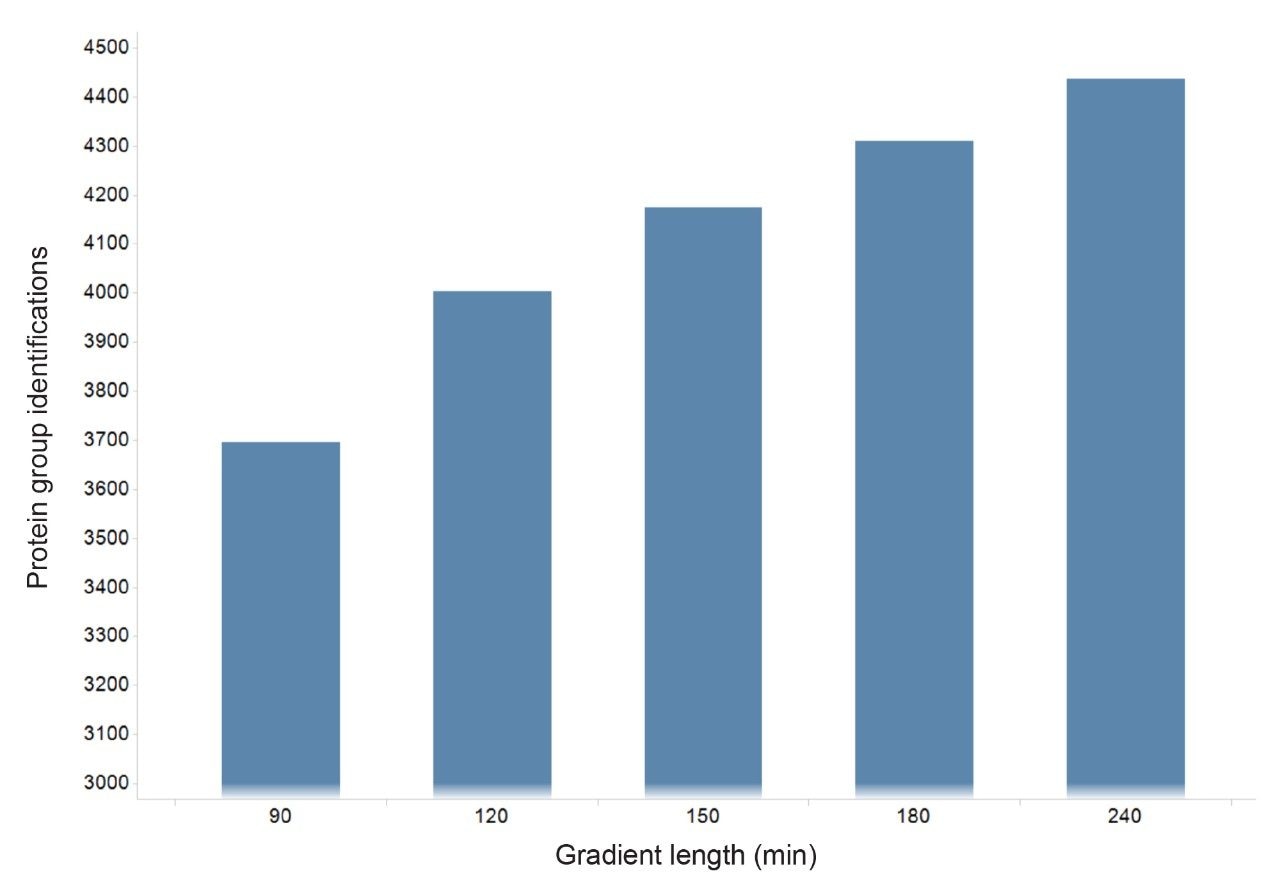 Figure 3. Protein identifications (FDR 1%) at different gradient lengths ranging from 90 to 240-minutes.
Figure 3. Protein identifications (FDR 1%) at different gradient lengths ranging from 90 to 240-minutes.
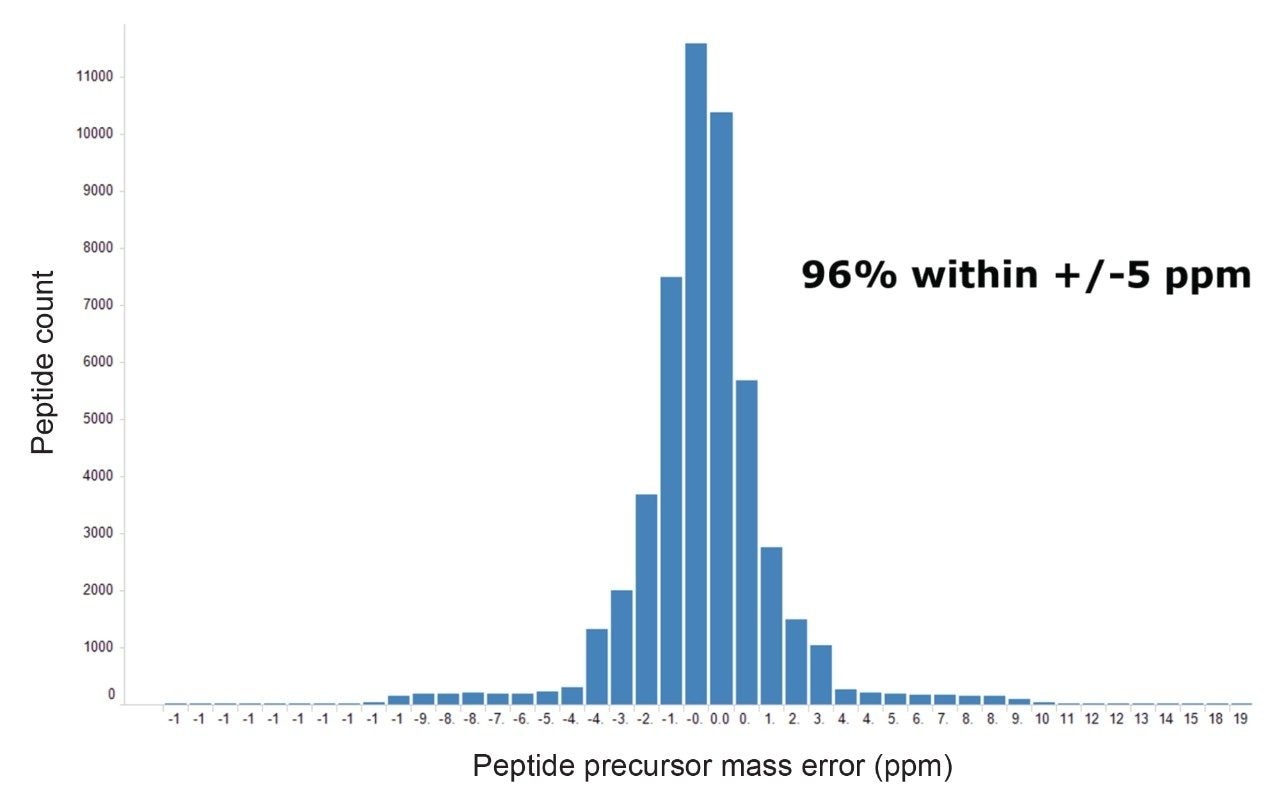 Figure 4. Distribution of mass accuracy (ppm) for all identified peptides relating to K562.
Figure 4. Distribution of mass accuracy (ppm) for all identified peptides relating to K562.
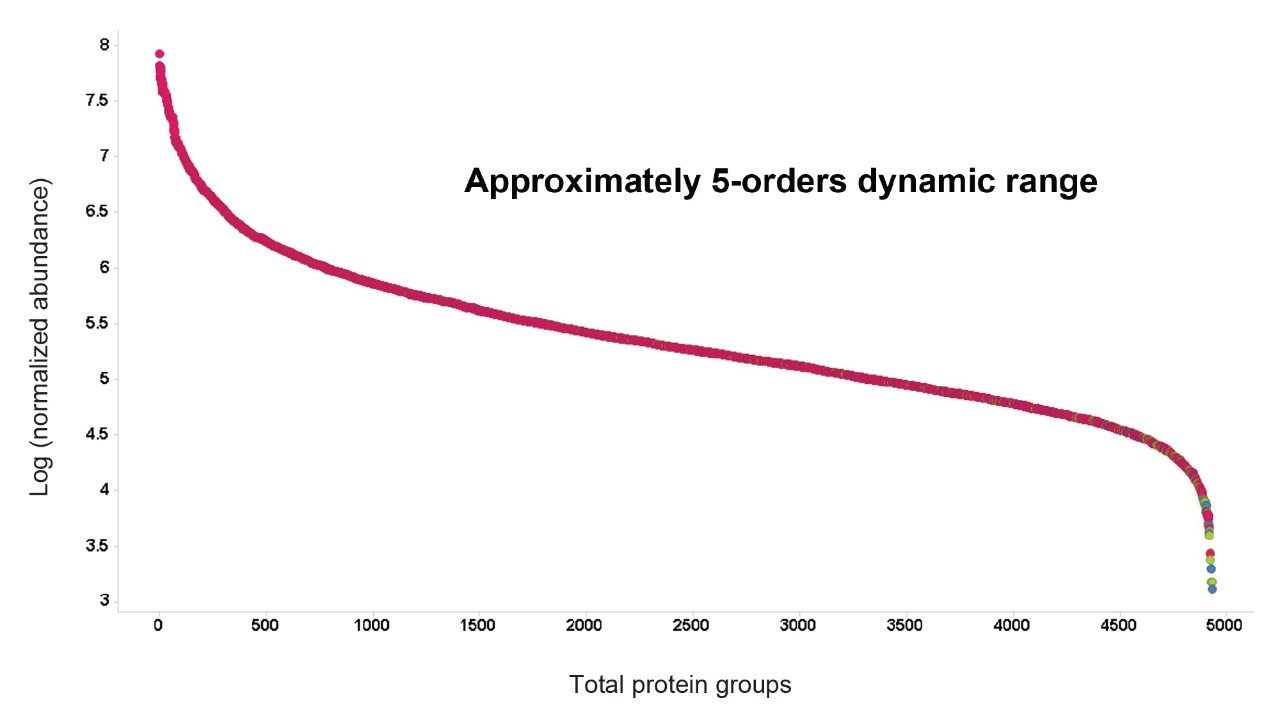 Figure 5. Dynamic range of intensities for all identified K562 proteins from 3 replicate injections.
Figure 5. Dynamic range of intensities for all identified K562 proteins from 3 replicate injections.
An important aspect for discovery experiments is injection to injection reproducibility, both in terms of signal intensity for quantitation purposes and quality of spectra for identification rates. These metrics are also an indication of the robustness of measurements achievable with the LC-MS system where some sample data acquisitions with tens or hundreds of samples can last several weeks. Figure 6 is a comparison between protein signal intensities of two replicate injections, specifically using the top 3 most intense peptides per protein from the search results. Figure 7 displays the identification rates for three technical analyses, establishing the number of identifications obtained in two out of the three injections. Based on this criteria, 3,602 proteins were identified with a 1% FDR, which equates to 78% of the total identifications. Similar findings based on the E. Coli results, provide an r2 value of 0.999 and 1,216 protein identifications between injections being obtained.
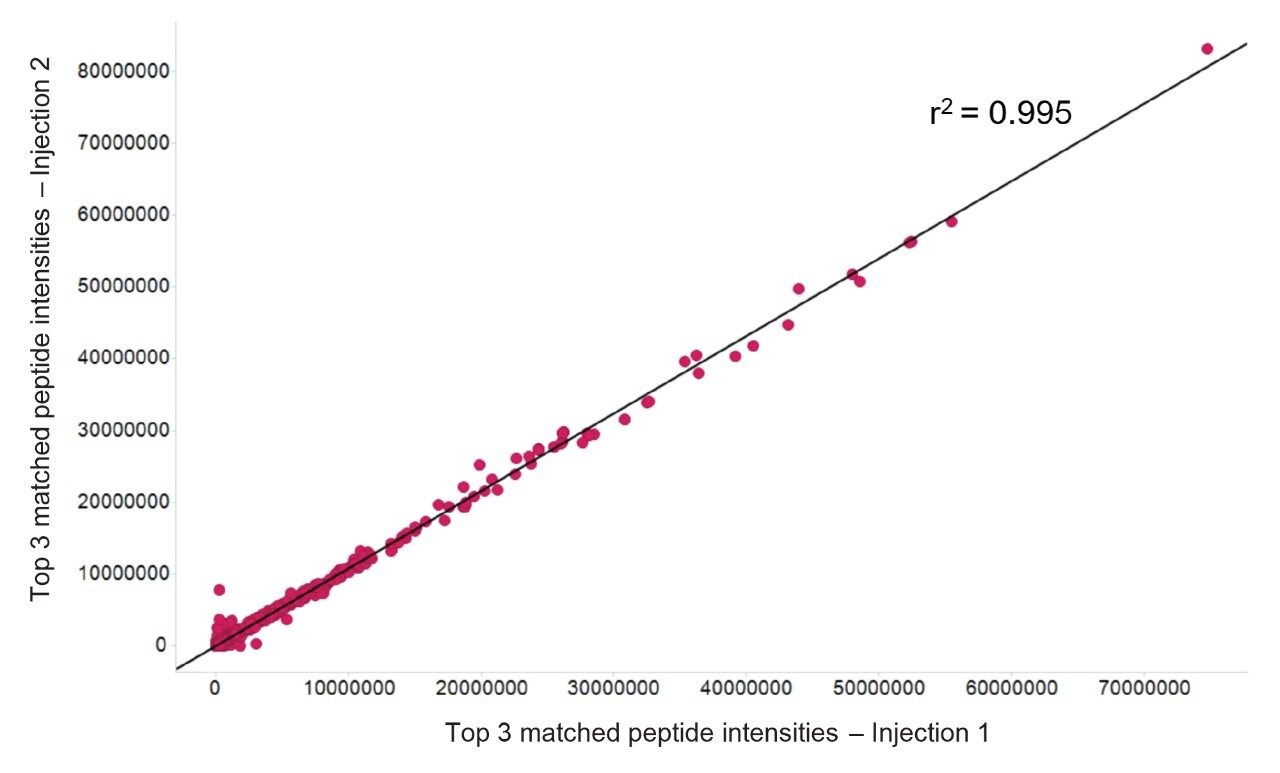 Figure 6. Injection to injection reproducibility for two replicate K562 injections, comparing the top 3 matched peptides per protein.
Figure 6. Injection to injection reproducibility for two replicate K562 injections, comparing the top 3 matched peptides per protein.
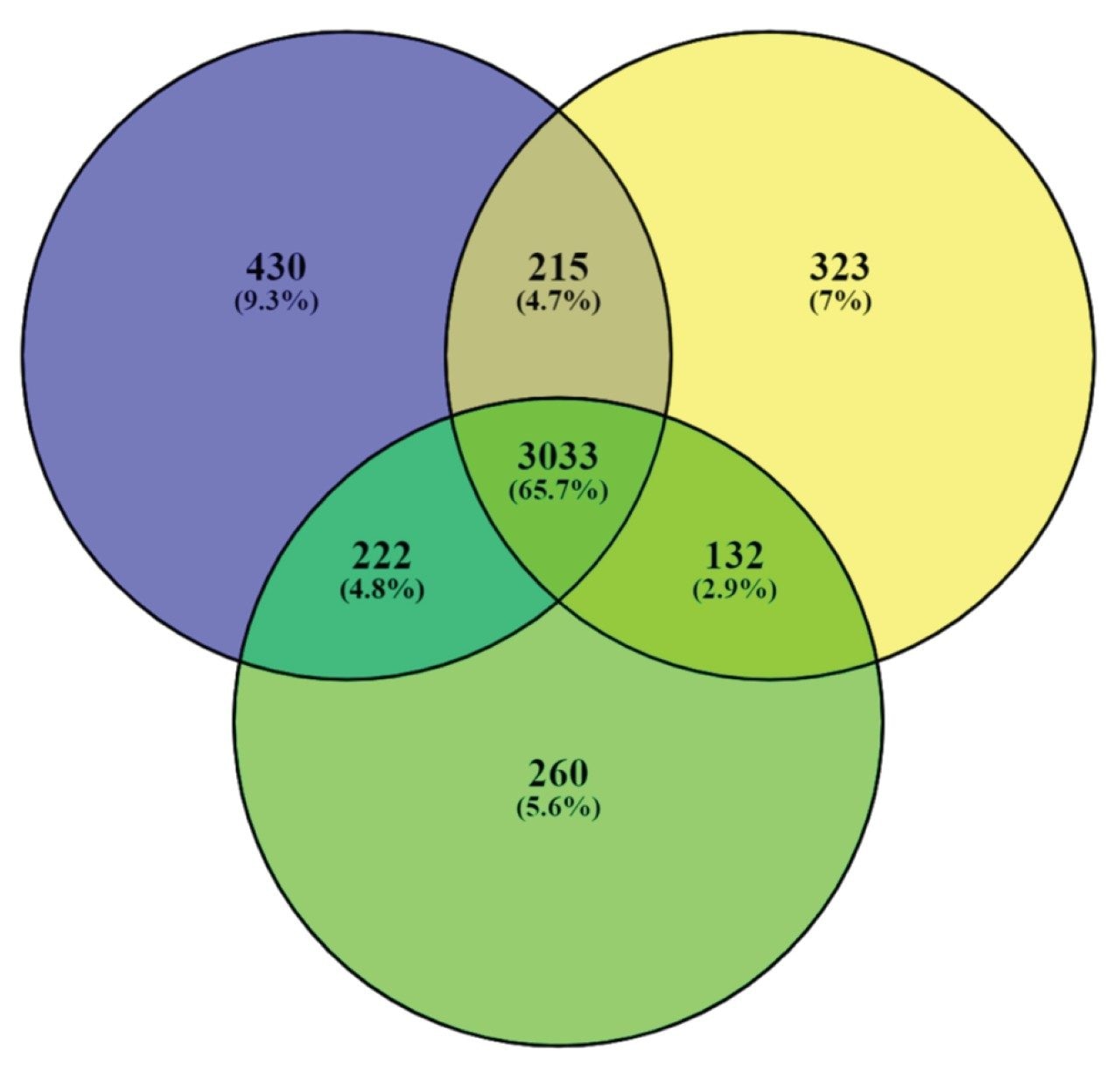 Figure 7. Venn diagram representing the overlap and reproducibility of protein identifications for three technical replicates. Based on two out of three replicates, resulted in 3,602 protein identifications (1% FDR) and 4,355 identifications based on 4% FDR.
Figure 7. Venn diagram representing the overlap and reproducibility of protein identifications for three technical replicates. Based on two out of three replicates, resulted in 3,602 protein identifications (1% FDR) and 4,355 identifications based on 4% FDR.
Quantitative performance is shown using Promega 6x5 peptides in E. Coli background matrix. The 6x5 mixture contains differentially stable isotope labelled peptides at varying levels, which all coelute from the LC column. Response from the differentially labelled series for one of the peptides, LASVSVSR, is shown in Figure 8. The data represents on column amounts ranging from 10 amol at the lowest level, up to 100 fmol with good linearity being obtained across the whole concentration range. Based on this example, we clearly demonstrate 5 logs of dynamic range.
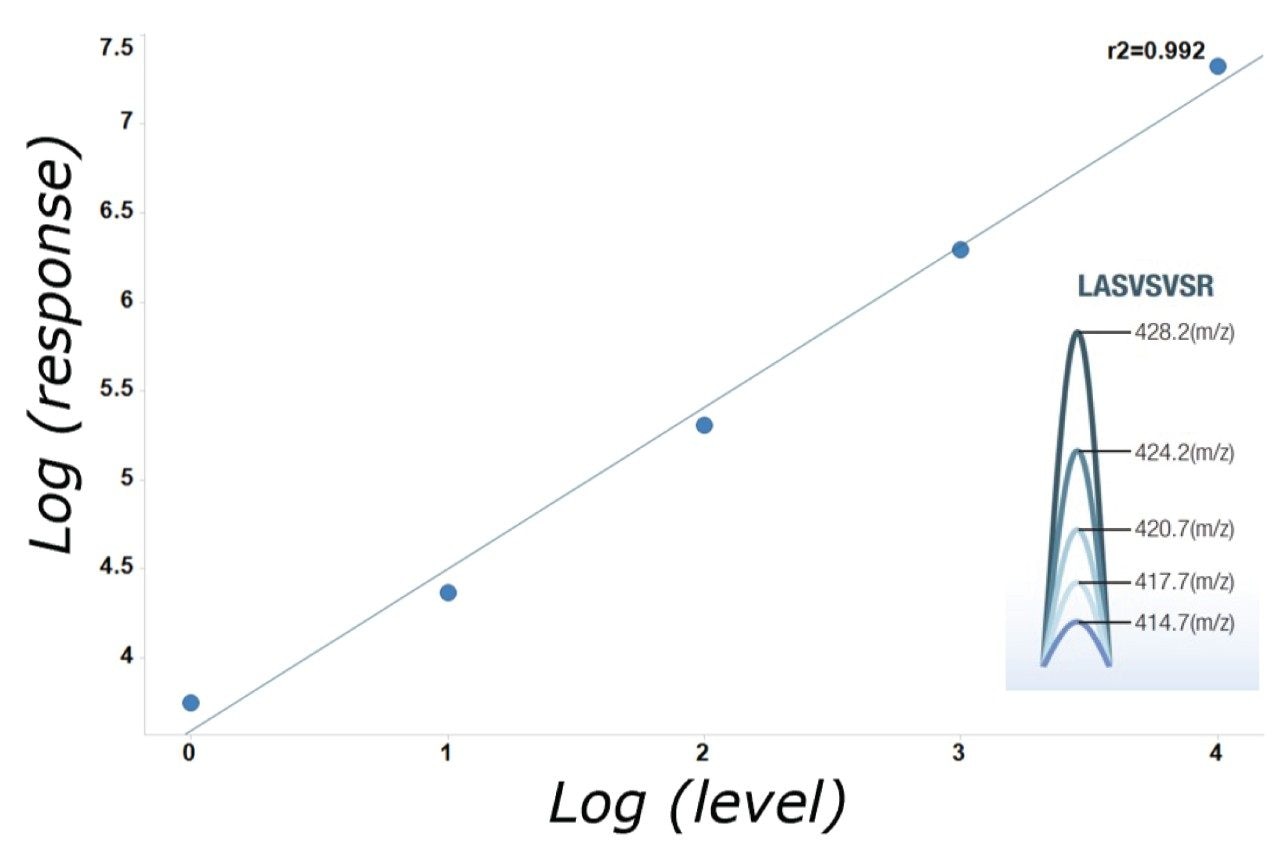 Figure 8. Linearity of peptide series based upon LASVSVSR spiked into an E. Coli background matrix. Data is representative of 10 amol to 100 fmol on column.
Figure 8. Linearity of peptide series based upon LASVSVSR spiked into an E. Coli background matrix. Data is representative of 10 amol to 100 fmol on column.
Conclusion
SELECT SERIES Cyclic IMS is the latest iteration of QTof Mass Spectrometers and when coupled to an ACQUITY UPLC M-Class at nanoscale flow rates, is shown to exhibit excellent performance for the proteomic analysis of medium to complex peptide digest mixtures. The data provided highlights not only the protein identification rates from individual experiments but also the excellent run to run reproducibility which is crucial to the successful analyses of large cohorts of samples. The enhanced dynamic range, up to 5 orders, of the system has been demonstrated by analyzing Human K562 peptide intensities from triplicate protein identifications and by using a differentially stable isotope labelled peptide mixture.
References
- Christopher J. Hughes, Lee A. Gethings. Characteristics of Proteomics Experiments Performed on the SYNAPT XS QTof Mass Spectrometer. Waters Application Note. 720006670EN, 2019.
Featured Products
720007381, September 2021