Efficient Non-Reduced mAb Subunit LC-MS Analysis to Screen for Modifications of Unpaired Cysteines in Innovator and Biosimilars
Abstract
This application note highlights a collaboration between Similis Bio and Waters™ Corporation to design a rapid screening method for the analysis of an unpaired cysteine modification in innovator and biosimilar monoclonal antibody (mAb) samples. The conserved sequences across IgG subclasses contain an even number of cysteine residues which are all paired in disulfide bonds.1 However, some mAbs contain an additional unpaired cysteine in the complementarity determining region (CDR) that may react with free cysteine or glutathione present in the mAb production process. Modification of these unpaired cysteines in the binding region may affect the biological activity of the mAb. The goal of the collaboration was to develop a simple and fast cysteine modification workflow based on liquid chromatography and mass spectrometry (LC-MS) to support large sample sets during process development of a biosimilar mAb at Similis Bio. A higher-throughput workflow to support this goal involved a mAb subunit digestion without a reduction step that would obfuscate the presence of an unpaired cysteine and cysteine modifications. Knowing this, we have developed a five-minute FabRICATOR® digestion protocol under non-reducing conditions, followed by five-minute analysis using the BioAccord™ LC-MS System. Using the INTACT Mass App within the waters_connect™ Informatics Platform, the resulting data was automatically charged deconvoluted, mass matched, and the relative modification abundance was calculated and reported for each sample as an injection finished acquisition.
Benefits
- Accelerated five-minute non-reduced FabRICATOR subunit mAb digestion and five-minute LC-MS analysis for rapid and unambiguous analysis of modifications on unpaired cysteines in the Fab region
- Streamlined data acquisition using BioAccord LC-MS System, a benchtop TOF MS designed for ease of use for analysts of all levels of expertise
- Utilization of the waters_connect Informatics Platform equipped with the Intact Mass App for automated near real-time data acquisition, analysis, and reporting with compliance-ready features
Introduction
Monoclonal antibodies (mAbs) have made up a major part of successful biopharmaceutical drug products over the past decade. With many of the older products approaching loss of market exclusivity, many companies are investing in the development of biosimilar mAb products that share the same protein sequences as the innovators but may exhibit differences in modifications due to the manufacturing cell line or process parameter differences. The fundamental goal for companies developing biosimilars is to prove that the biosimilar product has “no clinically meaningful differences from the reference product”.2 This means that any primary structure variants (e.g., deamidation, oxidation, etc.) that potentially impact the biological activity of the molecule must be present at similar levels when compared back to the reference product. Liquid chromatography coupled with mass spectrometry (LC-MS) is a powerful tool used to characterize these modifications at the intact, reduced subunit, and peptide level.
Another important modification, though less common in biopharmaceutical mAb products, is the reaction of any unpaired cysteine residues in the sequence. IgGs are comprised of two heavy chains (HC) and two light chains (LC) connected via interchain disulfide bridges between cysteine residues. Other cysteines in the heavy and light chains pair up via intrachain disulfide bridges. Both inter- and intra- chain disulfides contribute to the stable tertiary structure of the molecule. Some mAbs may contain additional cysteine residues in the hypervariable region which are unpaired. This has been shown to cause instability leading to aggregation, which then translated to a loss of biological activity.3 Even in the absence of stability issues, modification of these residues (e.g., cysteinylation, glutathionylation) during manufacturing may directly impact biological activity and compromise biosimilarity.
Modifications of unpaired cysteines at low yield present a unique challenge to typical LC-MS analysis on the intact mAb level. The mass shifts of cysteinylation and glutathionylation are +119 Da and +305 Da, respectively. For cysteinylation, in particular, the mass shift is very close to the C-terminal lysine variant (+128 Da) and will generally get lost amongst the complex set of N-glycoforms. These typical modifications are removable with enzymes such as carboxypeptidase B and PNGaseF, respectively, but the sample preparation may be longer, and processing may not be complete for some molecules. To circumvent the added complexity for intact mAb analysis, scientists may turn to mAb subunit workflows.4-7 However, unpaired cysteine modifications are difficult to analyze using these methods, as any addition of typical reducing agents will remove the modification and it will no longer be detectable in the analysis. Taking it a step further to analyze at the peptide mapping level brings the same challenge, as the additional sample preparation complexity and time requirements are not well suited to targeted analysis.
To achieve an efficient and rapid unpaired cysteine modification screening workflow, we opted to work toward a non-reduced mAb subunit analysis. Limited enzymatic digestion has been one approach historically used for liberating hinge derived mAb subunits, but tools such as the IdeS enzyme have become a more direct method of reliably generating mAb subunits for LC-MS analysis.8 The IdeS enzyme cleaves at a single amino acid site below the hinge. Under typical reducing conditions, this generates three roughly equal sized (~25 kDa) subunit species. The enzyme is also highly effective under non-reduced conditions (generating one ~100 kDa (Fd’+LC)2 subunit and two copies of the ~25 kDa Fc species). One of the most widely used IdeS enzymes on the market is FabRICATOR (Genovis AB). Pairing the efficiency and high specificity of FabRICATOR enzyme with a rapid UPLC™-TOF analysis on the BioAccord System, we have successfully implemented a subunit workflow to monitor modifications of unpaired cysteines (Figure 1). These results were confirmed, and site identification was made possible by non-reduced peptide mapping of the innovator sample using RapiZyme™ Trypsin.9
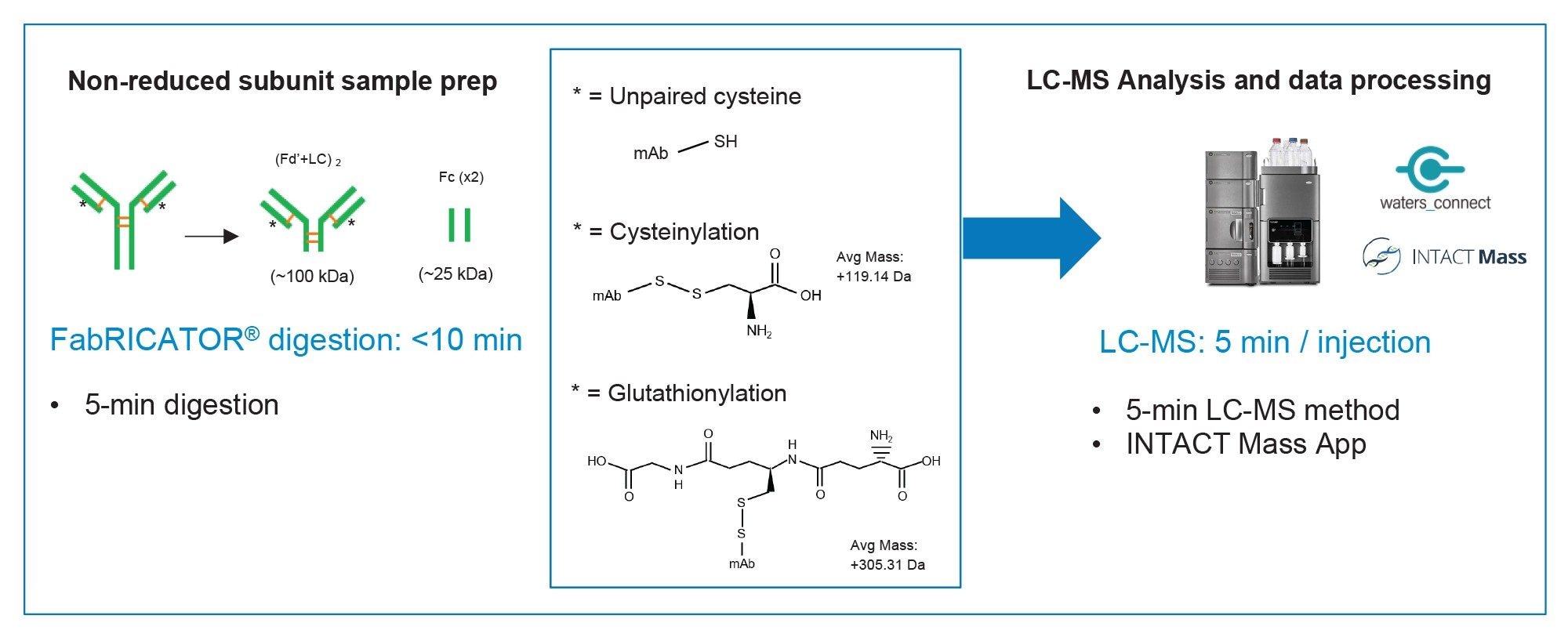 Figure 1. Overview of non-reduced subunit LC-MS workflow, from sample preparation with FabRICATOR to data acquisition and analysis with the BioAccord & waters_connect INTACT Mass App.
Figure 1. Overview of non-reduced subunit LC-MS workflow, from sample preparation with FabRICATOR to data acquisition and analysis with the BioAccord & waters_connect INTACT Mass App.
Experimental
Sample Preparation (Non-reduced Subunit): 3.3 µL of ~1.5 mg/mL each sample (biosimilar samples obtained directly from a bioreactor and purified using ProA capture) were added to 20 µL FabRICATOR Master Mix (0.5 units/µL FabRICATOR in 25mM Tris, pH 7.5) and incubated at 37 ⁰C for 5 minutes. (5 µg sample incubated with ten units FabRICATOR (1:2 ratio)). Samples were diluted 1:1 (v/v) with 0.1% formic acid in water for LC-MS analysis (0.1 mg/mL final sample concentration).
Sample Preparation (Non-reduced Peptide Mapping): 50 µg of innovator reference material was denatured, and free cysteines were alkylated in 6M Urea, 2.5 mM Iodoacetamide (IAM) for 30 minutes at 50 ⁰C (50 µL total, mAb concentration is at 1 mg/mL at this step). The sample was diluted 7x with Digestion Buffer (100 mM Tris HCl, 10 mM CaCl2, pH 7.5, p/n: 186010111), and 10 µg RapiZyme Trypsin (p/n: 186010108) was added (1:5 w/w). (Sample is at 0.14 mg/mL and 0.85M Urea after dilution.) The sample was digested for 2 hours at 37 ⁰C, and then quenched to a final 0.3% formic acid concentration (v:v).
mAb Subunit
LC Conditions
|
LC system: |
ACQUITY Premier UPLC System |
|
Detection: |
ACQUITY UPLC TUV (280 nm) |
|
Column(s): |
ACQUITY Premier BEH™ C4 300 Å, 1.7 µm, 2.1 x 50 mm (p/n: 186010326) |
|
Column temperature: |
80 oC |
|
Sample temperature: |
6 oC |
|
Injection: |
0.25 µg FabRICATOR-digested mAb (2.5 µL injection of 0.1 mg/mL sample) |
|
Flow rate: |
0.4 mL/min |
|
Mobile phase A: |
0.1% Formic Acid in Water |
|
Mobile phase B: |
0.1% Formic Acid in Acetonitrile |
Gradient Table
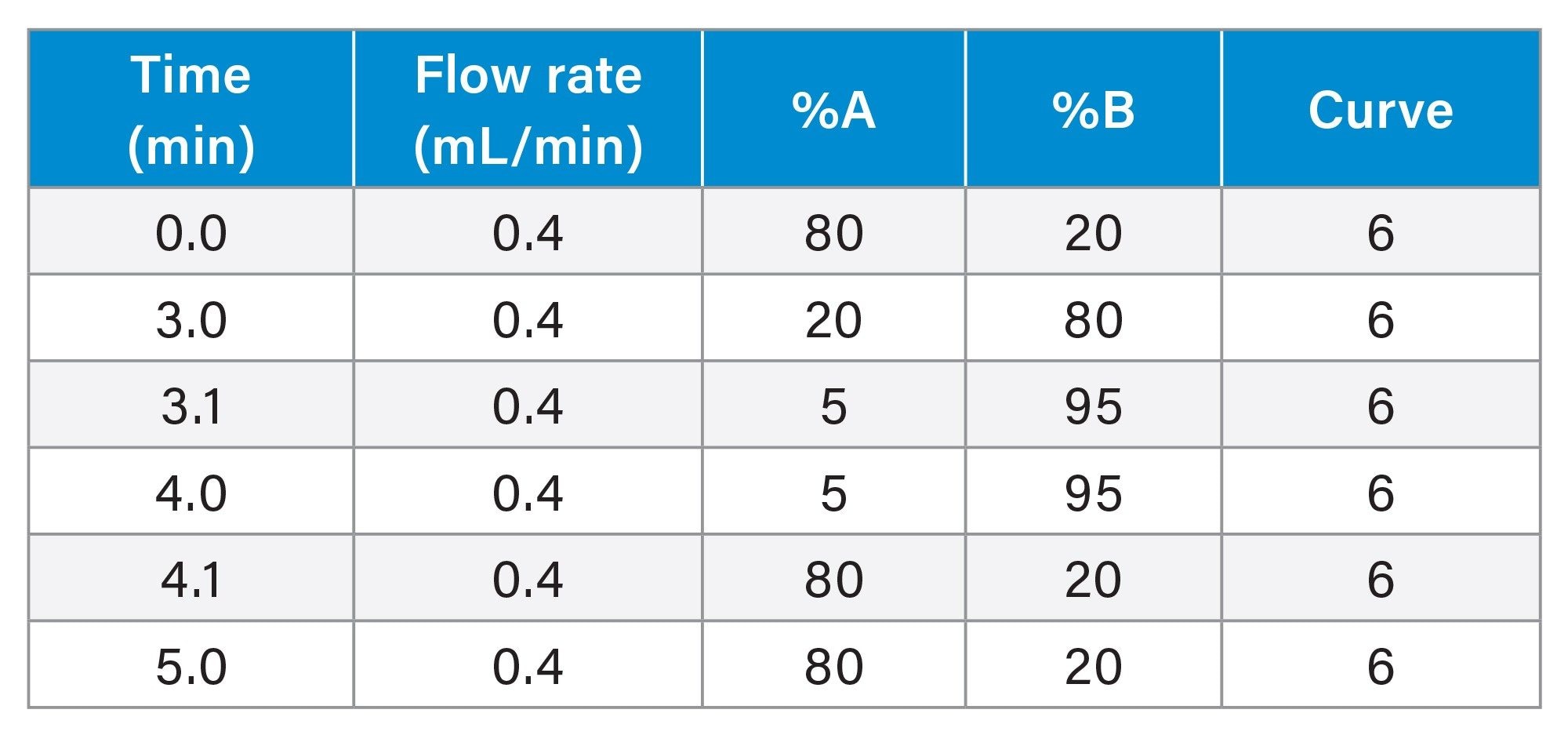
MS Conditions
|
MS System: |
ACQUITY RDa™ |
|
Ionization mode: |
ESI Positive, Full Scan |
|
Acquisition range: |
400–7000 m/z (High Mass) |
|
Capillary voltage: |
1.5 kV |
|
Cone voltage: |
50 V |
|
Desolvation temperature: |
550 °C |
|
Intelligent data capture: |
On |
|
Divert valve: |
Waste for 0–0.5 minutes and 4–5 minutes MS for 0.5–4 minutes |
Peptide Mapping- Focused DDA Experiment
*Note: A typical full-length DIA (MSE) based peptide mapping characterization method (outlined in detail in this application note, 720007840.9) was first used to identify peptides of interest. Once identified, the LC gradient was adapted to a short 15-minute method and DDA (data dependent acquisition) with a m/z inclusion list was utilized to characterize both the unmodified and low-level modified cysteinylation species.
LC Conditions
|
LC system: |
ACQUITY Premier UPLC System |
|
|
Column(s): |
ACQUITY Premier Peptide CSH™ C₁₈ 130 Å, 1.7 µm, 2.1 x 100 mm (p/n: 186009488) |
|
|
Column tempertaure: |
60 oC |
|
|
Sample temperature: |
6 oC |
|
|
Injection: |
0.2 µg non-reduced tryptic digest (1.5 µL injection of 0.14 mg/mL sample) |
|
|
Flow rate: |
0.2 mL/min |
|
|
Mobile phase A: |
0.1% Formic Acid in Water |
|
|
Mobile phase B: |
0.1% Formic Acid in Acetonitrile |
Gradient Table (Focused 15-minute method)
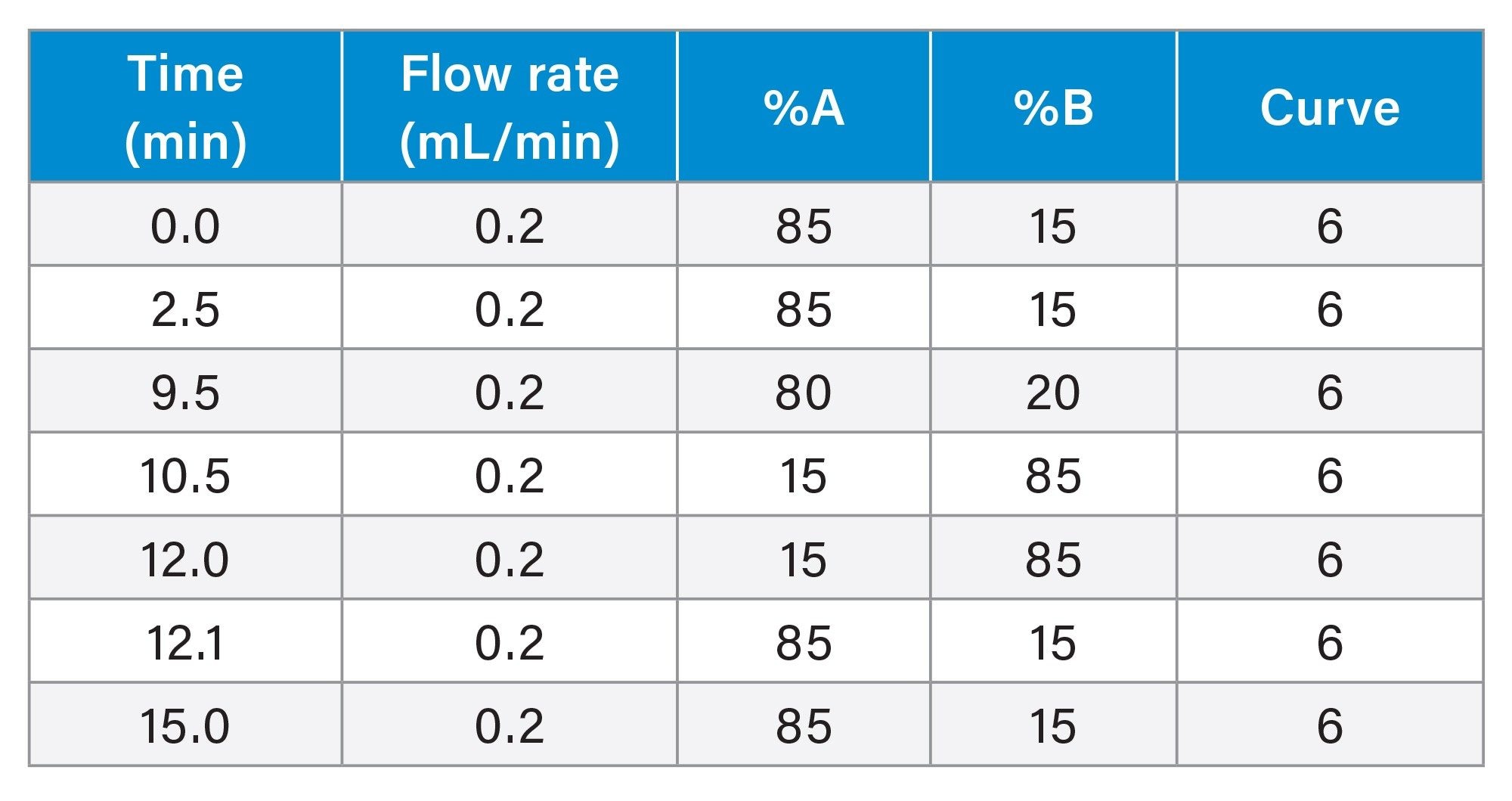
MS Conditions: Xevo™ G3 (Targeted DDA method for peptides of interest)
|
MS system: |
Xevo G3 QTof |
|
Ionization mode: |
ESI Positive, Sensitivity Mode |
|
Experiment type: |
DDA with Inclusion List |
|
Acquisition range: |
50–2000 m/z |
|
Capillary voltage: |
2.2 kV |
|
Cone voltage: |
20 V |
|
Source temperature: |
120 oC |
|
Desolvation temperature: |
350 oC |
|
Cone gas: |
35 L/h |
|
Desolvation gas: |
600 L/h |
|
Collision energy: |
Mass Dependent Ramp (Low Mass 10-30 V, High Mass 25-55 V) |
|
DDA settings: |
• Scan Time 0.2 seconds • MSMS: Maximum two simultaneous MSMS acquisitions • Use Survey Scan Settings • Start MSMS when intensity exceeds 7500 counts/seconds • Stop MSMS until Condition (Cumulative TIC rises above threshold 3,000,000 counts) or Timeout (5.0 seconds) • Charge States: 2+, 3+, 4+, 5+ • Deisotope: Off |
|
DDA Inclusion List: |
1. Peak at RT: m/z 913.39 (4+ charge state) at 8.1 min 2. Peak at RT: m/z 928.89 (4+ charge state) at 7.4 min |
Data Management
Subunit data was acquired and processed through the Intact Mass App (v 1.4.0.0) within the waters_connect Informatics Platform (v 3.1). Peptide mapping data was collected through the UNIFI™ App (v 3.1.0.16) within the waters_connect Informatics Platform (v 3.1) and processed by the Peptide Mapping workflow.
Results and Discussion
The non-reduced mAb subunit workflow implemented here offers a path forward for rapid screening of unpaired cysteine residues and their potential modifications. In this study, it was applied to an innovator and subset of biosimilar samples provided by Similis Bio to assess the impact of various cell culture conditions on the levels of cysteinylation and glutathionylation, with the end-goal of quickly processing large sample sets to support biosimilar process development. After the samples were prepared in a five-minute non-reducing FabRICATOR digestion, they were analyzed using a five-minute LC-MS method on the BioAccord LC-MS System. With the use of the Acquire and Process function in the INTACT Mass App, scientists were able to quickly submit samples for analysis, monitor real-time data progress, and view automatically deconvoluted and reported results as each injection, as they finished acquisition.
Under non-reducing conditions, the FabRICATOR enzyme yielded a covalently linked (Fd’+LC)2 species (~100 kDa) and disassociated Fc chains (~25 kDa), as observed in the UV (Figure 2A) and TIC (Figure 2B) chromatograms. The Fc species (Peak 1) contained the N-glycoforms and unprocessed C-terminal lysine which would obscure the cysteinylation and glutathionylation results if intact mAb analysis was performed. With the Fc modification complexity removed, the (Fd’+LC)2 species (Peak 3) then only showed possible glycation, unpaired cysteine modifications, and/or oxidation. (Note: isomerization and deamidation are also possible for the (Fd’+LC)2 species, but due to their small mass difference they are only discernable using peptide level analysis.) Another added benefit of this non-reduced FabRICATOR method is that both arms of the Fab domain remain intact, and therefore, both the singly and doubly modified unpaired cysteine species were present and quantifiable. This information may prove critical, especially if there are differences in biological activity based on if one or both cysteines are modified, especially in multivalent antibodies.
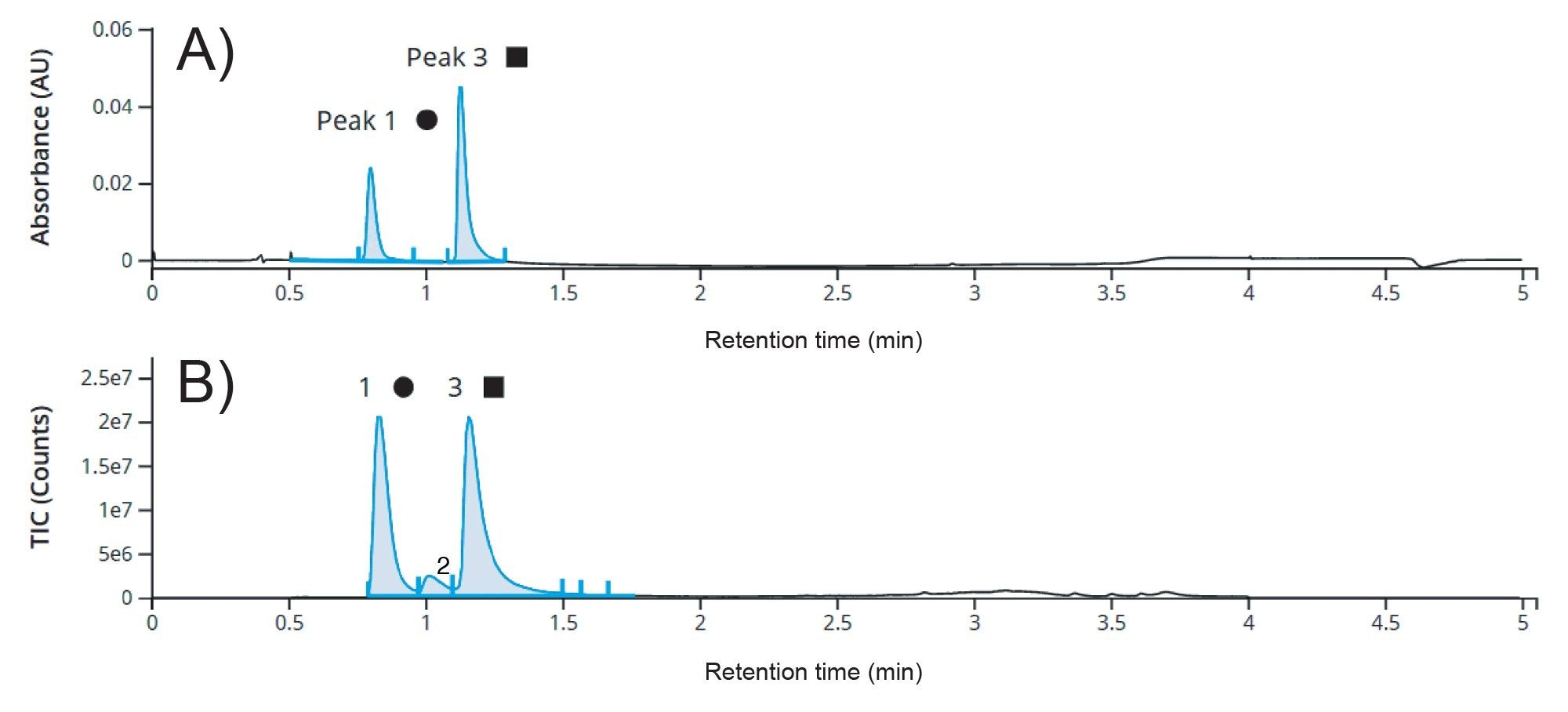 Figure 2. UV (Panel A) and TIC (Panel B) chromatograms for 0.25 µg injection of non-reduced FabRICATOR digest of the innovator mAb in a five-minute LC-MS analysis. Peak 1 corresponds to the Fc (~25 kDa) species and Peak 3 corresponds to the (Fd’+LC)2 (~100 kDa) species. (The small peak (#2) has a mass corresponding to the FabRICATOR enzyme.)
Figure 2. UV (Panel A) and TIC (Panel B) chromatograms for 0.25 µg injection of non-reduced FabRICATOR digest of the innovator mAb in a five-minute LC-MS analysis. Peak 1 corresponds to the Fc (~25 kDa) species and Peak 3 corresponds to the (Fd’+LC)2 (~100 kDa) species. (The small peak (#2) has a mass corresponding to the FabRICATOR enzyme.)
The INTACT Mass App processing method included variable modifications for cysteinylation, glutathionylation, glycation, and oxidation, which were automatically assigned to the deconvoluted masses based on a maximum mass match error of 20 ppm. The percentage relative abundance was calculated for all assigned forms of the (Fd’+LC)2 species and reported in the summary table for each analysis. The deconvoluted spectra (Figure 3) are displayed for a subset of the samples—the innovator with “low” level of cysteine modification, plus two biosimilar samples containing a “medium” and “high” level of cysteine modification. The +1 and +2 cysteinylations are noted with red arrows, and the +1 glutathionylation is noted with green arrows (+2 glutathionylation was not detected for any of the samples). The detailed results for the sample set are reported in Table 1. The ~5% modification on the innovator is in line with the expected value based on orthogonal data provided by Similis Bio. The biosimilar samples were observed with cysteine modifications in the range of 16-63%. Glycation of the (Fd’+LC)2 species was measured in the range of 2-11% for this sample set. This subunit mAb method provided confident relative quantitation of individual species down to 0.5% based on these results.
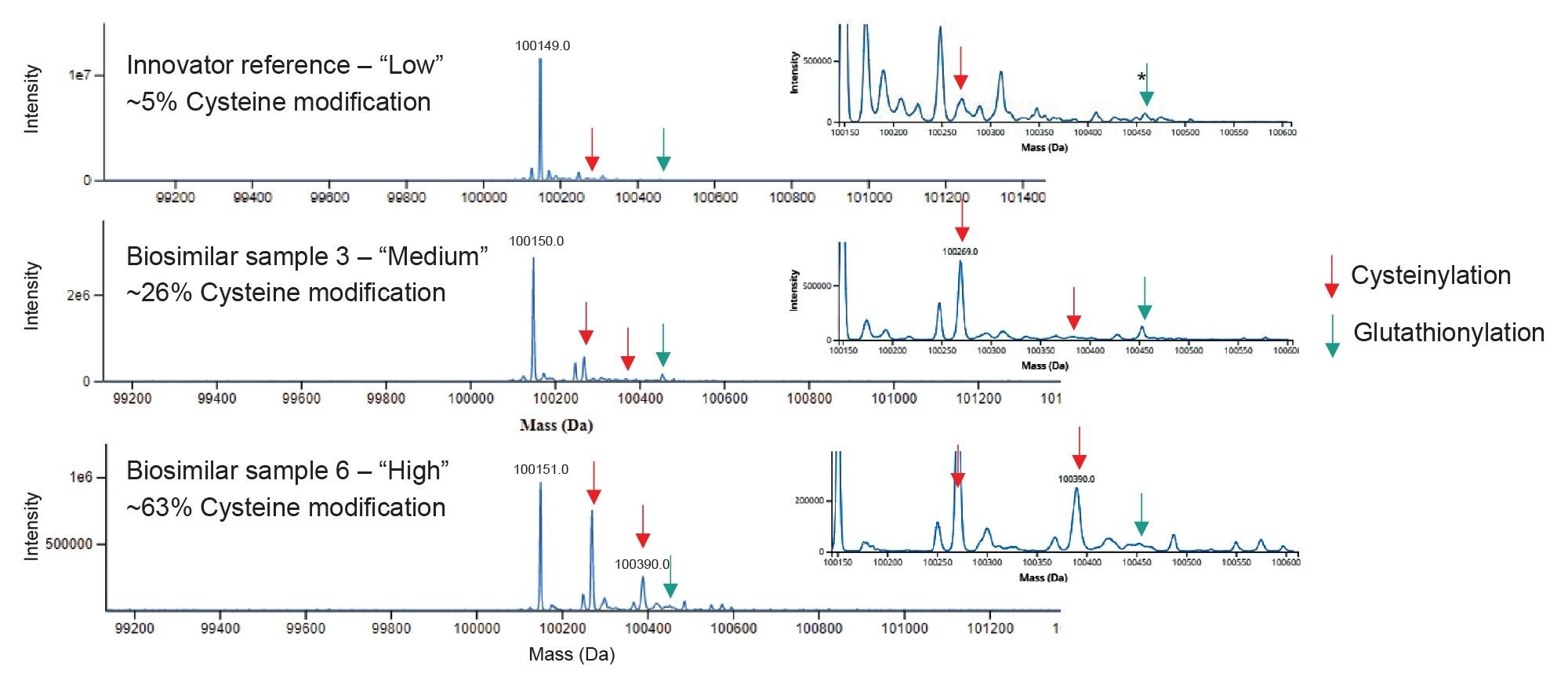 Figure 3. Deconvoluted spectra for (Fd’+LC)2 species of non-reduced FabRICATOR digestions for the Innovator Reference, Biosimilar Sample 3, and Biosimilar Sample 6, with “low”, “medium”, and “high” levels of cysteine modification, respectively, with a zoomed section showing the modifications. Deconvoluted via Auto Deconvolution setting in the Intact Mass App. *Low level glutathionylation assignment for the innovator is supported by the confident assignment in biosimilar samples with elevated levels.
Figure 3. Deconvoluted spectra for (Fd’+LC)2 species of non-reduced FabRICATOR digestions for the Innovator Reference, Biosimilar Sample 3, and Biosimilar Sample 6, with “low”, “medium”, and “high” levels of cysteine modification, respectively, with a zoomed section showing the modifications. Deconvoluted via Auto Deconvolution setting in the Intact Mass App. *Low level glutathionylation assignment for the innovator is supported by the confident assignment in biosimilar samples with elevated levels.
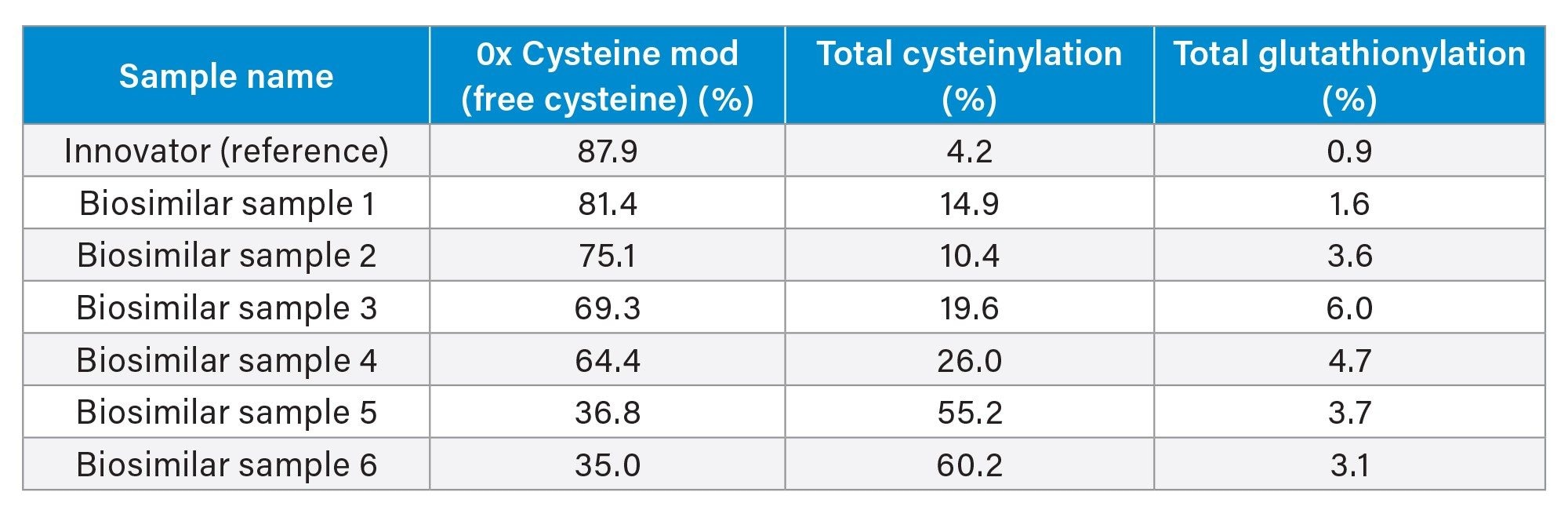 Table 1. Summary of unpaired cysteine modification results for Innovator and Biosimilar samples originating from various manufacturing processes. The “Total” cysteinylation and glutathionylation values are a sum of the (Fd’+LC)2 species with one unpaired cysteine modified and both unpaired cysteines modified. The remaining 2 - 11% of (Fd’+LC)2 species deconvoluted MS signal from glycated species.
Table 1. Summary of unpaired cysteine modification results for Innovator and Biosimilar samples originating from various manufacturing processes. The “Total” cysteinylation and glutathionylation values are a sum of the (Fd’+LC)2 species with one unpaired cysteine modified and both unpaired cysteines modified. The remaining 2 - 11% of (Fd’+LC)2 species deconvoluted MS signal from glycated species.
To confirm that the cysteinylation observed in the innovator sample was indeed located on the expected unpaired cysteine in the light chain, non-reduced peptide mapping analysis was performed. The sample was denatured with urea and pre-alkylated with IAM to block any unpaired cysteines present prior to digestion with trypsin. If unpaired cysteines are not blocked with an alkylating reagent, they are more prone to disulfide scrambling during digestion. This complicates the analysis, as it creates false disulfide-bridged peptide species. Then the sample was diluted to reduce the urea concentration for RapiZyme Trypsin digestion using a 1:5 w/w enzyme:protein ratio. The resulting digest was quenched and analyzed with a previously published LC-MS method using the Xevo G3 QTof Mass Spectrometer operating in DIA (MSE) fragmentation mode.10
The peptide containing the assumed unpaired cysteine also contains a second cysteine residue which is involved in an intrachain disulfide bridge. Thus, the species of interest was actually two peptides connected via disulfide bridge, labeled “2:T2–2:T7”. This peptide species has either the +1 cysteinylation modification or the +1 “IAM” (pre-alkylation via iodoacetamide), meaning that this was the unpaired, unmodified cysteine. These two species elute at 26.5 and 27.2 minutes, respectively, as shown in the integrated TIC chromatogram (Figure 4).
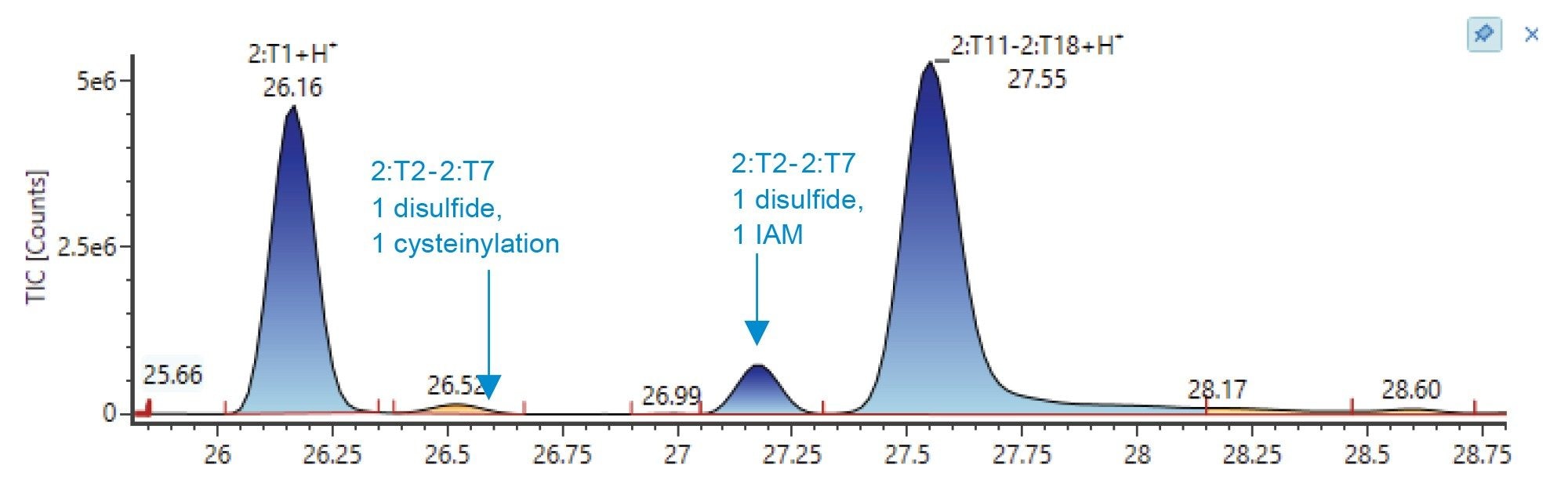 Figure 4. Non-reducing peptide mapping results. This figure displays a zoomed section of the integrated TIC chromatogram where the peptides containing the expected unpaired cysteine elute. The cysteinylated species co-eluted with another peptide in the TIC chromatogram (26.5 min) but is clearly distinguishable in the resulting deconvoluted MS spectra.
Figure 4. Non-reducing peptide mapping results. This figure displays a zoomed section of the integrated TIC chromatogram where the peptides containing the expected unpaired cysteine elute. The cysteinylated species co-eluted with another peptide in the TIC chromatogram (26.5 min) but is clearly distinguishable in the resulting deconvoluted MS spectra.
Once the peptides of interest were detected in the longer LC gradient method, a short 15-minute LC-MS method with a focused gradient was developed for a targeted analysis (data not shown). In the 15-minute method the modified cysteine ( m/z 928.9) and unmodified cysteine ( m/z 913.4) peptide species eluted at 7.4 and 8.1 minutes, respectively. This retention time and m/z information was used to create a targeted DDA (data dependent acquisition) with an inclusion list to ensure the detection and fragmentation of only these peptides of interest. The resulting fragmentation data (Figure 5) from the cysteinylated and alkylated peptide species were compared to determine the location of the modification. The presence of y9 + cysteine in the cysteinylated peptide species (and corresponding y9 + IAM alkylation in the unpaired cysteine peptide species) confirms the location as the expected unpaired cysteine (highlighted in yellow).
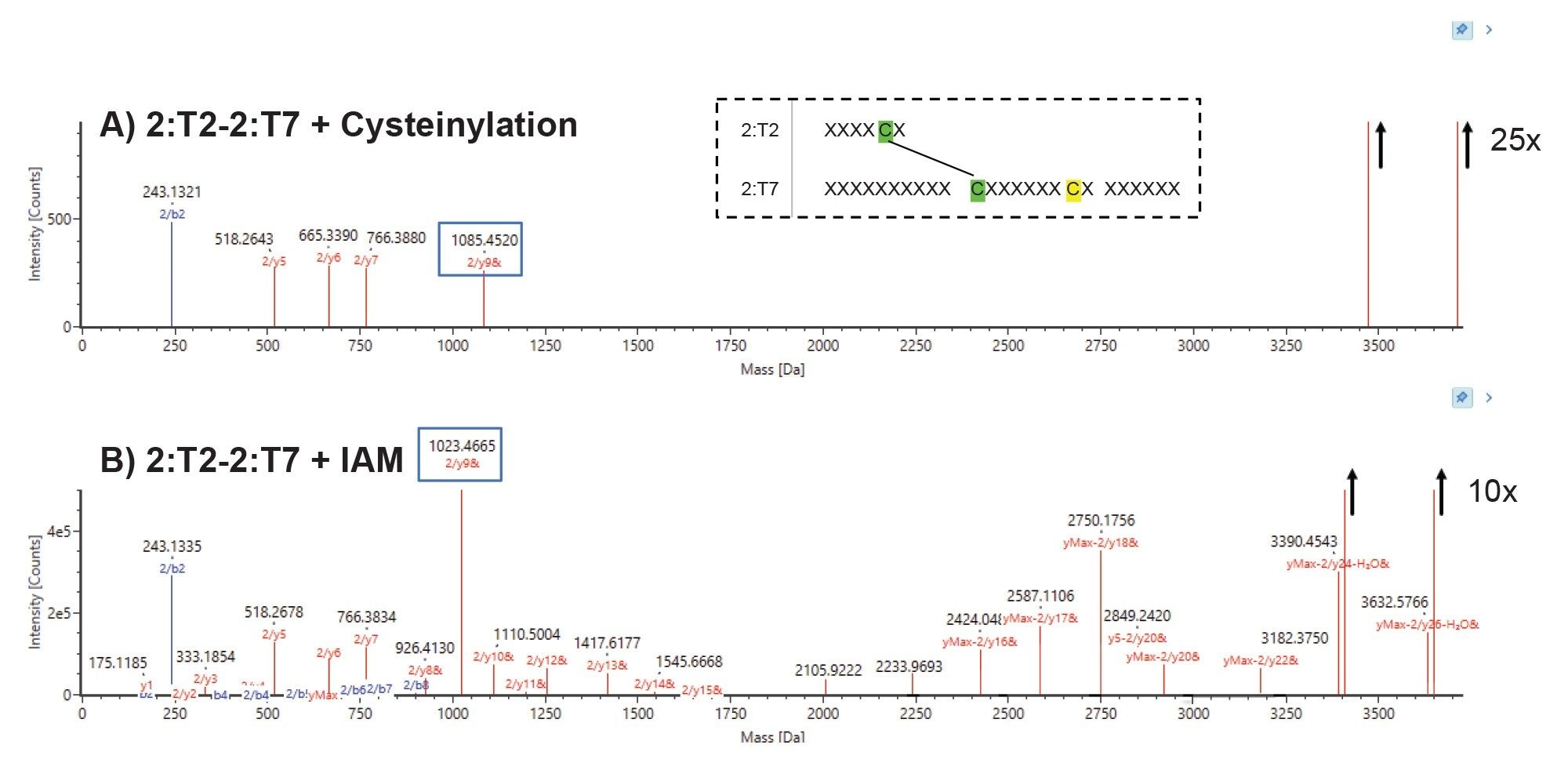 Figure 5. Fragmentation map of matched fragments from the disulfide linked peptides (2:T2–2:T7) containing the expected unpaired cysteine (highlighted in yellow). Panel A: cysteinylation modified and Panel B: alkylated form with IAM. The location of the cysteinylation was confirmed to be on the expected unpaired cysteine through the examination of y fragment ladder, with the modification mass difference series beginning at the cysteine corresponding to the y9 fragment and continuing through the y-max fragment.
Figure 5. Fragmentation map of matched fragments from the disulfide linked peptides (2:T2–2:T7) containing the expected unpaired cysteine (highlighted in yellow). Panel A: cysteinylation modified and Panel B: alkylated form with IAM. The location of the cysteinylation was confirmed to be on the expected unpaired cysteine through the examination of y fragment ladder, with the modification mass difference series beginning at the cysteine corresponding to the y9 fragment and continuing through the y-max fragment.
Conclusion
This application note demonstrates a successful method optimization of a rapid non-reduced subunit mAb screening workflow to measure levels of cysteinylation and glutathionylation on unpaired cysteines in the Fd’ or LC of IgG1 mAbs. The workflow utilized a five-minute FabRICATOR digestion under non-reducing conditions, followed by LC-MS analysis via a BioAccord LC-MS System operated under the compliance-ready waters_connect Informatics Platform. Utilization of the INTACT Mass App automated deconvolution, mass assignment, and reporting capabilities provided a streamlined workflow for going from sample to report. In this case, the study of innovator and biosimilar mAb samples from Similis Bio, a wide range (5–63%) of cysteine modification levels was observed using the non-reduced subunit method, with the unpaired modified cysteine residue confirmed on the predicted site by a non-reduced peptide mapping of the innovator reference sample.
This optimized subunit mAb LC-MS workflow for free and modified cysteine site analysis can be easily adopted and implemented to support product and process development for innovator or biosimilar candidates. The use of the waters_connect informatics with the BioAccord System provides the ability to deploy this same platform in support of manufacturing or product quality assessments, should the site prove to be a critical quality or process attribute.
References
- Liu H, May K. Disulfide bond structures of IgG molecules: Structural Variations, Chemical Modifications and Possible Impacts to Stability and Biological Function. mAbs. 2012; 4(1): 17–23.
- Biosimilar Basics for Patients. FDA website. <https://www.fda.gov/drugs/biosimilars/biosimilar-basics-patients> Accessed 7 Nov 2023.
- Banks DD, Gadgil HS, Pipes GD, Bondarenko PV, Hobbs V, Scavezze JL, Kim J, Jiang X, Mukku V, Dillon TM. Removal of Cysteinylation from an Unpaired Sulfhydryl in the Variable Region of a Recombinant Monoclonal IgG1 Antibody Improves Homogeneity, Stability, and Biological Activity. J Pharm Sci. 2008; 97:2, 775–790.
- Ippoliti S, Yu YQ, Ranbaduge N, Chen W. Establishment of a Robust mAb Subunit Product Quality Attribute Monitoring Method Suitable for Development, Process Monitoring, and QC Release. Waters Application Note 70007129. 2021.
- Sokolowska I, Mo J, Dong J, Lewis M, Hu P. Subunit Mass Analysis for Monitoring Antibody Oxidation. mAbs. 2017; 9:3, 498–505.
- Sokolowska I, Mo J, Pirkolachahi F, McVean C, Meijer L, Switzar L, Balog C, Lewis M, Hu P. Implementation of a High-Resolution Liquid Chromatography-Mass Spectrometry Method in Quality Control Laboratories for Release and Stability Testing of a Commercial Antibody Product. Anal Chem. 2020; 92, 2369–2373.
- Nägeli A, Ekemohn M, Nyhlén H. Automated Middle-level Analysis of Therapeutic mAbs in Complex Protein Samples. Genovis Application Note. AN0056.
- Gadgil HS, Bondarenko PV, Pipes GD, Dillon TM, Banks D, Abel J, Kleemann GR, Treuheit MJ. Identification of Cysteinylation of a free Cysteine in the Fab Region of a Recombinant Monoclonal IgG1 Antibody Using Lys-C Limited Proteolysis Coupled with LC/MS Analysis. Anal Biochem. 2006; 355, 165–174.
- Ippoliti S, Zampa N, Yu YQ, Lauber MA. Versatile and Rapid Digestion Protocols for Biopharmaceutical Characterization Using RapiZyme Trypsin. Waters Application Note 720007840. 2023.
- DeLaney K, Ippoliti S, Reid L, Cornwell O, Yu YQ, Harry E, Towers M. Applying Peptide Mapping and Multi-Attribute Method (MAM) Workflow for Biosimilar mAb Drug Products Comparison on the Xevo G3 QTof Platform. Waters Application Note. 720007632. 2022.
FabRICATOR is a registered trademark of Genovis AB. BioAccord, RapiZyme, waters_connect, ACQUITY, BEH, CSH, and RDa are trademarks of Waters Technologies Corporation.
720008305, April 2024