RNA CQA Analysis using the BioAccord LC-MS System and INTACT Mass waters_connect Application
For research use only. Not for use in diagnostic procedures.
Abstract
This application note highlights new informatics capabilities to analyze the critical quality attributes (CQAs) of ribonucleic acid (RNA) based therapeutics. Ion-Pair Reversed Phase (IP-RP) Liquid Chromatography-Mass Spectrometry (LC-MS) data was acquired using the BioAccord™ LC-MS System under the control of the waters_connect™ informatics platform. Application specific software has been developed including the mRNA Cleaver microApp (to generate in silico fragments), the INTACT Mass App (to detect and assign RNA digestion products), and the Coverage Viewer microApp (to visualize digestion data coverage). This workflow was demonstrated for CQAs of both single guide RNAs (sgRNAs) and messenger RNAs (mRNAs), typical of the latest generation of therapeutics and vaccines.
Benefits
- Data analysis workflow for RNA digestion fragment mapping, the mass confirmation of nucleic acid digestion products, using a new version of the INTACT Mass App for data processing and additional microApp tools for in silico generation and curation of digest fragments
- Additional workflows using the INTACT Mass App for measuring mRNA 5’ Capping efficiency and 3’ poly(A) tail heterogeneity
Introduction
There has been a surge in the development of RNA-based therapeutics and vaccines in the last decade.1-3 The recent development and approval of the two COVID mRNA-based vaccines has brought RNA-based therapeutics to the forefront of the biopharma industry.4-6 As such, development of analytical methods for monitoring the CQAs of RNA-based therapeutics has become a high priority for ensuring proper control of manufacturing processes. CQAs for nucleic acid therapeutics can include sequence confirmation, 5’ capping efficiency (mRNA) and structure, 3’ poly(A) tail heterogeneity (mRNA) analysis, localization of modified nucleotides, and purity assessment of the active RNA product.
LC-MS systems have been established as an effective investigative tool when characterizing CQAs of RNA-based therapeutics.7-10 However, data processing is a common bottleneck that needs to be addressed. Within this application note, we present a data processing workflow for RNA CQA analysis using tools developed on the waters_connect informatics platform, specifically the INTACT Mass, mRNA cleaver, and Coverage Viewer applications.
Experimental
Reagents and Sample Preparation
The experimental conditions reported here are for the sgRNA digestion and sequence mapping experiments. The appropriate experimental details and sample information for the mRNA CQA data within this application note can be found in the following Waters application notes: mRNA mapping data (Gaye et al), 3’ poly(A) Tail data (Doneanu et al), and the 5’ cap data (Ngyuen et al).13,14,18
Data processing workflow example:
N,N-diisopropylethylamine (DIPEA, 99.5% purity, catalogue number 387649-100ML), 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, 99% purity, catalogue number 105228-100G), ethanol (HPLC grade, catalogue number 459828-2L) and ammonium bicarbonate (LiChropur LC-MS Supelco reagent, catalogue number 5330050050) were purchased from Millipore Sigma (St Louis, MO). Acetonitrile (LC-MS grade, catalogue number 34881-1L) and methanol (LC-MS grade, catalogue number 34966-1L) were obtained from Honeywell (Charlotte, NC). HPLC grade Type I deionized (DI) water was purified using a Milli-Q system (Millipore, Bedford, MA). Mobile phases were prepared fresh daily. Ultrapure nuclease-free water (catalogue number J71786.AE) for sgRNA digestions was purchased from Thermo Fisher Scientific (Waltham, MA).
100-mer sgRNA was purchased from IDT (Integrated DNA Technologies, Coralville, IA) and reconstituted in 100 µL of nuclease-free water. The sgRNA contained six stability-enhancing modified nucleotides at terminal positions 1, 2, 3, and 97, 98, 99 in the sequence (Figure 1A). The modified nucleotides included were 2’O-methylguanosine, 2’O-methyluridine, and 2’O-methyladenosine and each modification had a thioated phosphate group (Figure 1B). RNase T1 ribonuclease was purchased from Worthington Biochemical Corporation (Lakewood, NJ). For sgRNA digestion, 5 µL of reconstituted sgRNA were mixed with 1 µL digestion buffer (100 mM ammonium bicarbonate), 43 µL of nuclease-free water and 1 µL of the 1 mg/mL RNase T1 solution (prepared in nuclease-free water). The digestion mixture was prepared in a QuanRecovery™ MaxPeak™ 300 µL vial and incubated at 37 °C for 15 minutes, then immediately analyzed by LC-MS.
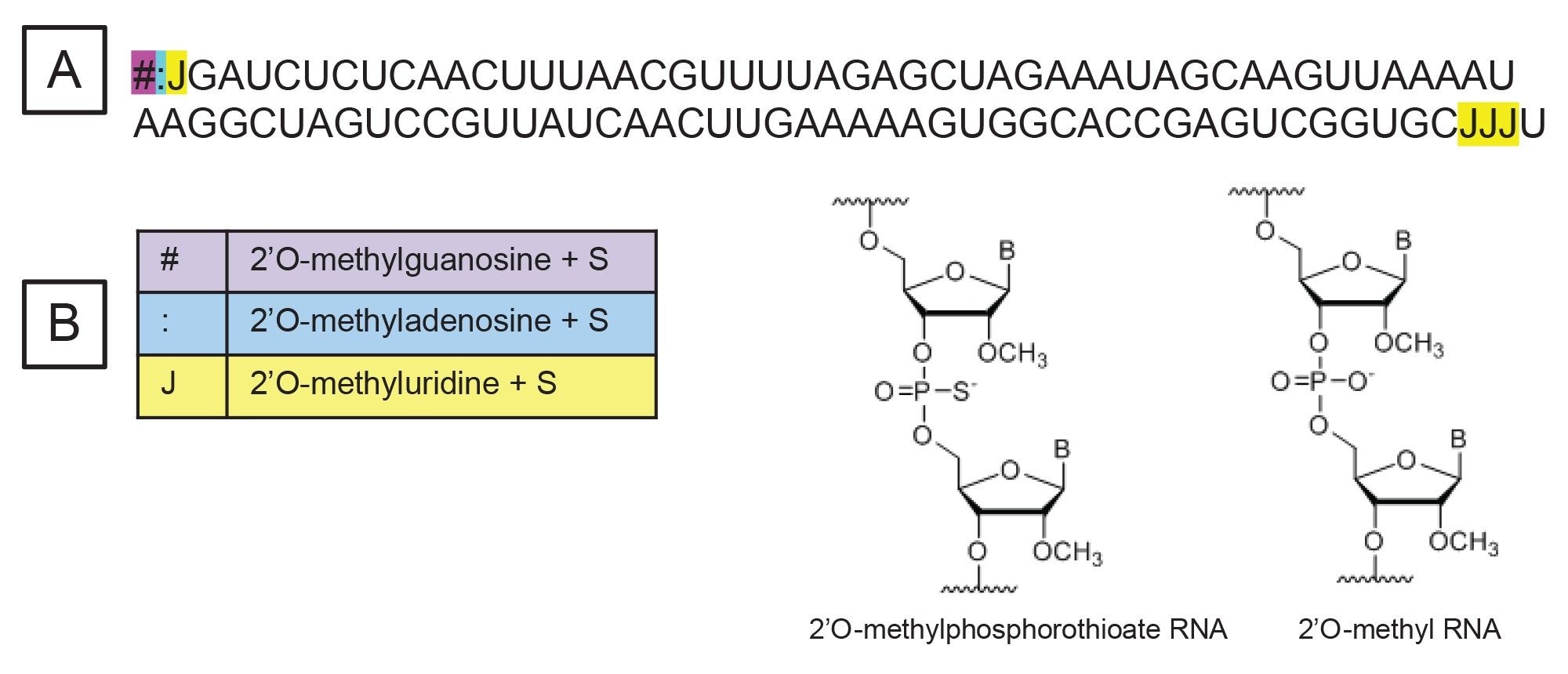 Figure 1. The sequence of the sgRNA analyzed. The ‘#’ represents 2’O-methylguanosine, the ‘:’ represents 2’O-methyladenosine, and the ‘J’ represents 2’O-methyluridine. All the modified nucleotides included a thioated phosphate group.
Figure 1. The sequence of the sgRNA analyzed. The ‘#’ represents 2’O-methylguanosine, the ‘:’ represents 2’O-methyladenosine, and the ‘J’ represents 2’O-methyluridine. All the modified nucleotides included a thioated phosphate group.
All datasets were acquired within the waters_connect informatics platform (version 3.2.0) using the UNIFI™ application (version 3.6.0) and processed using the INTACT Mass application (version 1.6.0). Versions 1.0.0 of the mRNA Cleaver and 1.0.0 of the Coverage Viewer microApps were used.
LC Conditions
|
LC-MS System: |
BioAccord LC-MS System with ACQUITY™ Premier UPLC™ (Binary) |
|
Pre-Column: |
VanGuard FIT cartridge holder (p/n: 186007949) containing a 2.1 x 5 mm ACQUITY Premier FIT cartridge packed with 1.7 µm BEH™ C18 particles. (p/n: 186009459) |
|
Column: |
ACQUITY Premier OST Column 1.7 µm, 130 Å, 2.1 x 150 mm, p/n: 186009486 |
|
Column temperature: |
60 °C |
|
Flow rate: |
300 µL/min |
|
Mobile phases: Solvent A: |
8 mM DIPEA (N,N-diisopropylethylamine), 40 mM HFIP (1,1,1,3,3,3-hexafluoroisopropanol), in DI water, pH 8.8 |
|
Solvent B: |
4 mM DIPEA, 4 mM HFIP in 75% ethanol |
|
Sample temperature: |
6 °C |
|
Sample vials: |
QuanRecovery MaxPeak HPS vials (p/n: 186009186) |
|
Injection volume: |
5 µL |
Gradient Table for sgRNA experiments
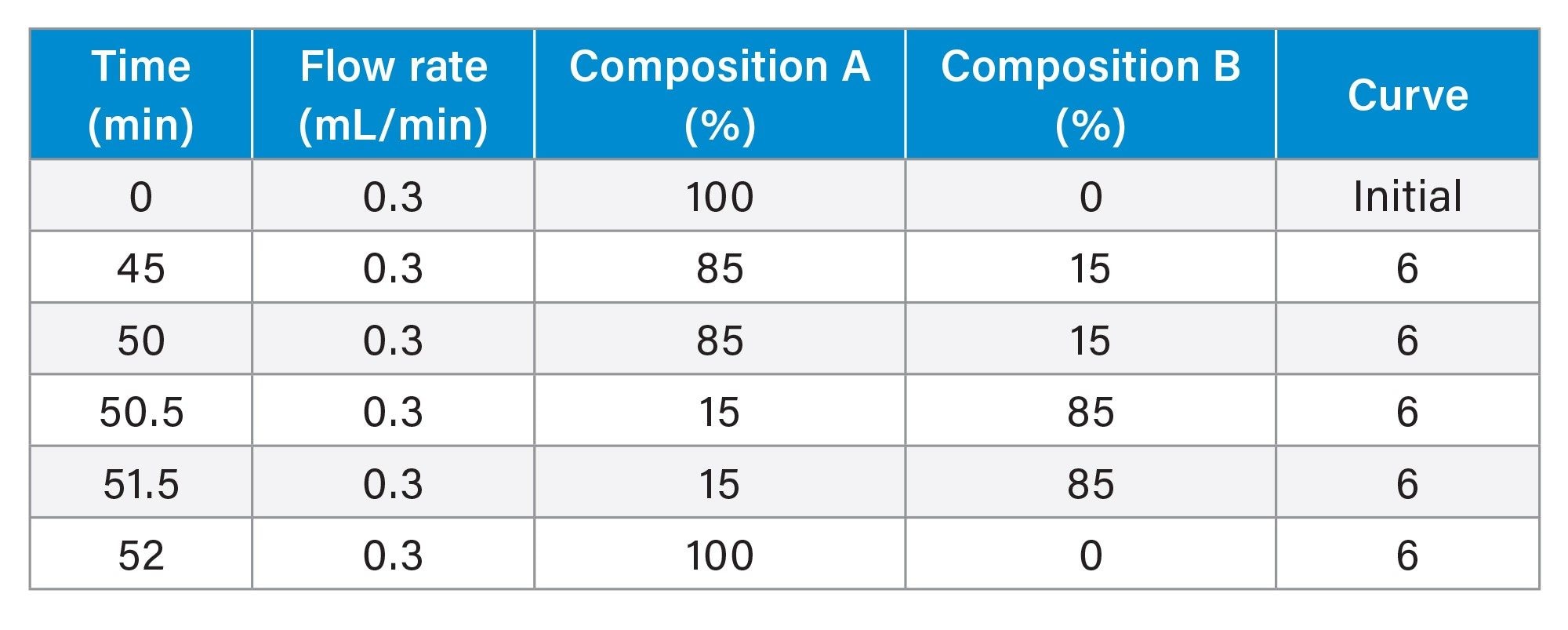
MS Conditions
|
Acquisition mode: |
Full scan |
|
Ionization mode: |
ESI (-) |
|
Capillary voltage: |
0.8 kV |
|
Cone voltage: |
45 V |
|
Source temperature: |
120 °C |
|
Desolvation temperature: |
500 °C |
|
Desolvation gas (N2) pressure: |
6.5 bar |
|
TOF mass range: |
400-5000 m/z |
|
Acquisition rate: |
2 Hz |
|
Lock-mass: |
waters_connect lockmass solution (p/n: 186009298) |
|
Informatics platform for data acquisition and processing: |
waters_connect v.3.2.0 |
|
Data acquisition: |
UNIFI App ver 3.6.0 |
|
Data processing: |
INTACT Mass App ver 1.6.0 |
Results and Discussion
Data Processing Workflow for Oligo Mapping
LC-MS based oligo mapping techniques have been developed over the past decades to address the growing need to analyze nucleic acid CQAs.19-23 Oligo mapping is analogous to peptide mapping as it provides the primary structure (the sequence) of the target nucleic acid through enzymatic digestion and accurate mass determination. Common enzymes used in oligo mapping include RNase T1, RNase A, and MazF. For the enzymatic digestions described here, we used RNase T1 which cuts the oligonucleotide after every guanosine on the 3’ end. The mechanism of RNase T1 has been studied extensively and described previously.24-27 As we only used one enzyme, we did not expect 100% sequence coverage. Typically, an oligo mapping experiment would use multiple enzymes in parallel as each enzyme has a different specificity and would produce a different set of digestion fragments. By doing so, the resulting digestion fragments from each enzyme should overlap in sequence, providing better sequence coverage. However, to keep this application note concise, we limited our scope to one enzyme.
Another tactic in oligo mapping experiments is to intentionally digest the oligonucleotide partially by altering enzymatic digestion conditions. By using partial digestion, the nucleic acid digestion fragments would be longer on average and therefore, more unique in both sequence and mass. While this approach can improve coverage, partial digestion can be hard to control, adding complexity to an already complex sample through additional, possibly overlapping m/z peaks and coeluting incomplete digestion fragments. For the data shown here, our intention was to completely digest the RNA in question, and we used enzymatic conditions optimized to do so.
1. Generating in silico digestion components
The data processing workflow for oligo mapping and other nucleic acid CQAs is illustrated in Figure 2. The generation of the in silico digestion fragments was accomplished using the mRNA Cleaver microApp. The Graphical User Interface (GUI) of the microApp is shown as Figure 3A. The User Guide is located on the left-hand side of the GUI to improve the user experience with the software. The mRNA Cleaver microApp is a flexible digestion calculator and library generator for oligo mapping investigations.
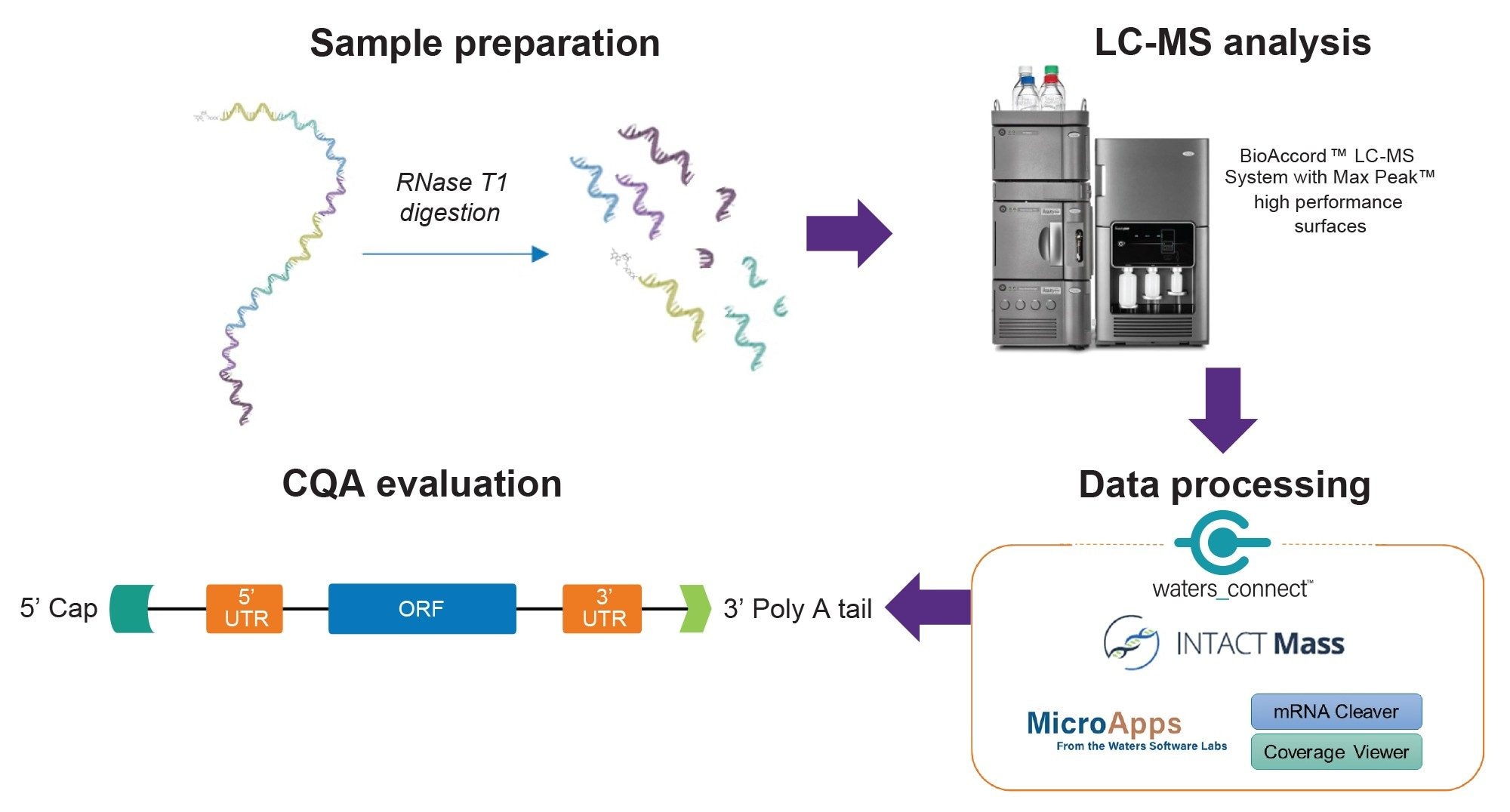 Figure 2. Workflow scheme for the characterization of RNA CQAs.
Figure 2. Workflow scheme for the characterization of RNA CQAs.
A user can modify three embedded files to customize the calculator to their specific needs. For example, if a sequence of interest contains modified nucleotides, the masses and chemical compositions can be added, as well as a representative single letter or symbol, to the “residues.csv” file, whose file path can be found in the User Guide. Using the representative letter or symbol of the modified nucleotide will prompt the program to use the adjusted masses and chemical composition to calculate the relevant digestion products. The “enzymes.csv” and “modifications.csv” files can similarly be modified to add different enzymes or mRNA-specific modifications (such as different 5’ caps or 3’ poly(A) distributions) to fit the user’s purpose.
Digestion specific settings are located below the sequence field (Figure 3A) and include adjusting the digestion product termini, whether a-specific digestion products should be generated, the number of missed cleavages to include, and whether mRNA specific modifications should be considered. Filters for the generated digestion product list are located on the right-hand side, where the generated digestion products are filtered based upon length settings. Additionally, the user can choose whether or not to check the box "Filter Duplicate Sequences". Duplicate sequences are digestion fragments that have the same sequence identity. By default, mRNA Cleaver will have that box checked which means that if there is a duplicate sequence it will be listed once with the alternate locations for that sequence listed in another column. For our workflow, the box is unchecked for use in Coverage Viewer in a subsequent data analysis step. MS specific settings are located below the filters, where the user can indicate the polarity of the expected mass ions, whether the generated list should reference monoisotopic or average masses, the charge state range to be generated for each digestion product, and the m/z range to be generated. The digestion product list is generated when the user clicks “Calculate”, and the list is output as a comma delimited file (.csv). An example output is shown in Figure 3B. This .csv file will be imported to the INTACT Mass as targeted search components processing.
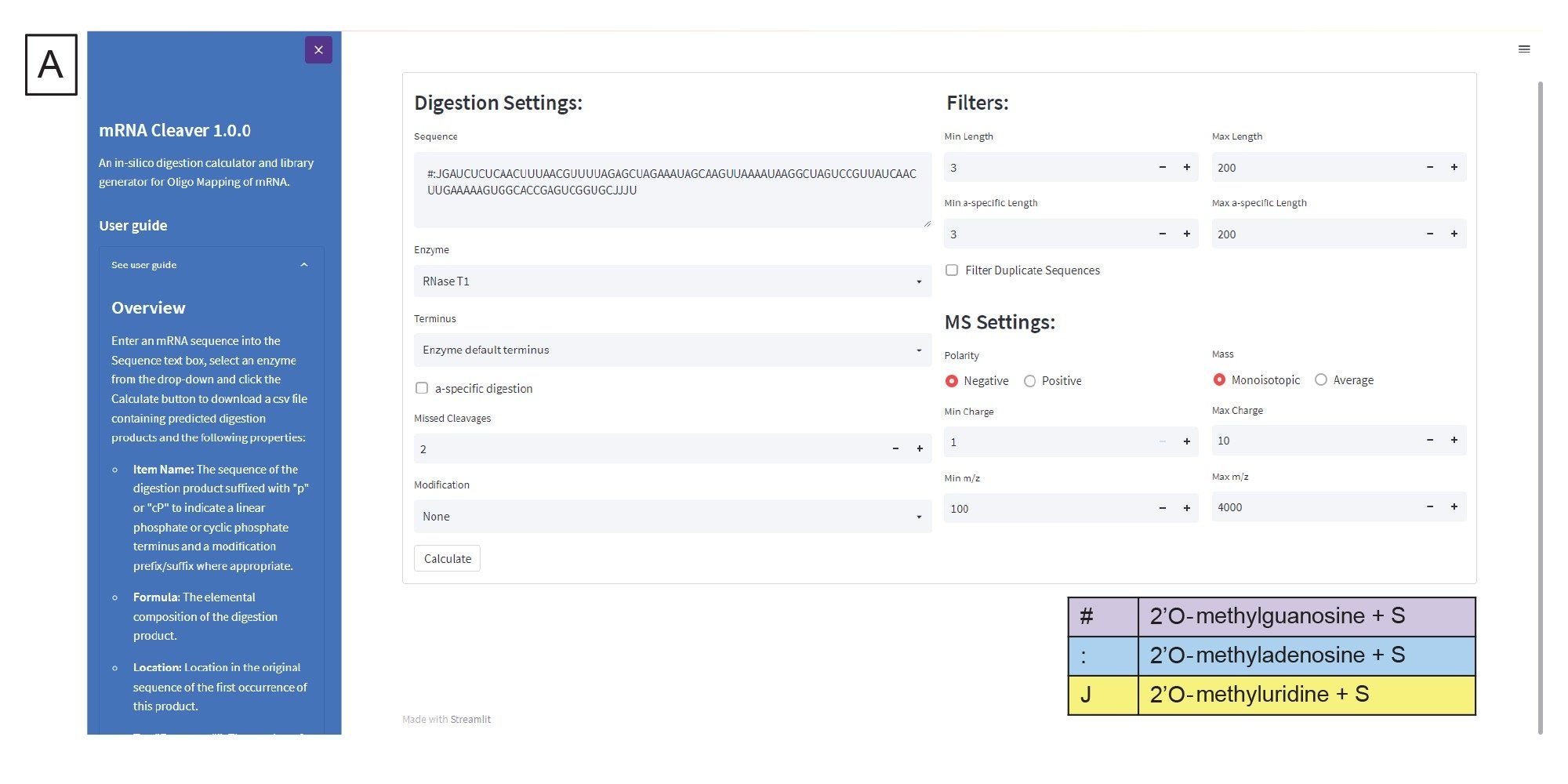 Figure 3. (A) The mRNA Cleaver microApp User Interface (GUI).
Figure 3. (A) The mRNA Cleaver microApp User Interface (GUI).
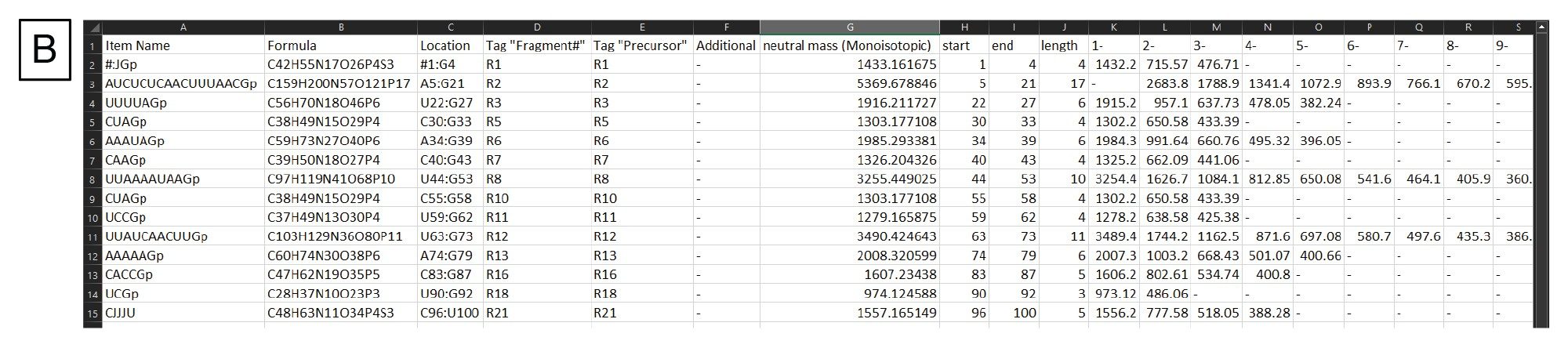 Figure 3. (B) Example output from mRNA Cleaver.
Figure 3. (B) Example output from mRNA Cleaver.
2. Oligo Fragment Mass Confirmation
Once the in silico digestion product list is generated and the experimental data is collected on the digested sample, the next step in the workflow is to process the data through the INTACT Mass App within waters_connect. The output from the mRNA Cleaver microApp provides the expected masses of the digestion products and their molecule IDs which can be copied directly into the “Expected Masses” column within the application. Modifiers can be added to include common adducts and sequence modifiers. Figure 4A contains the results screen after the analysis has finished processing while Figure 4B illustrates a representative deconvoluted spectrum output. The INTACT Mass results, MS, UV, and TIC tables can be downloaded using the download button on the right-hand side of the table.
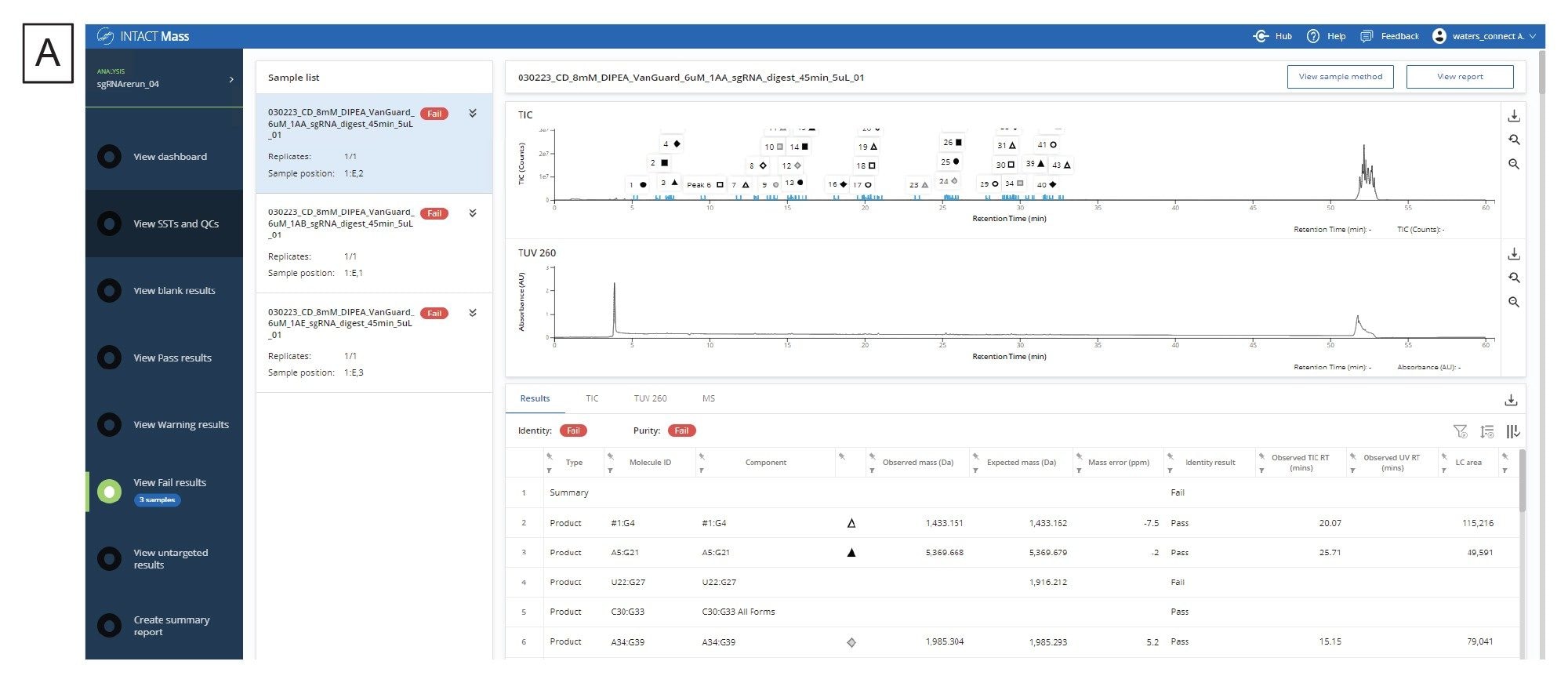 Figure 4. (A) The results dashboard of the INTACT Mass Application.
Figure 4. (A) The results dashboard of the INTACT Mass Application.
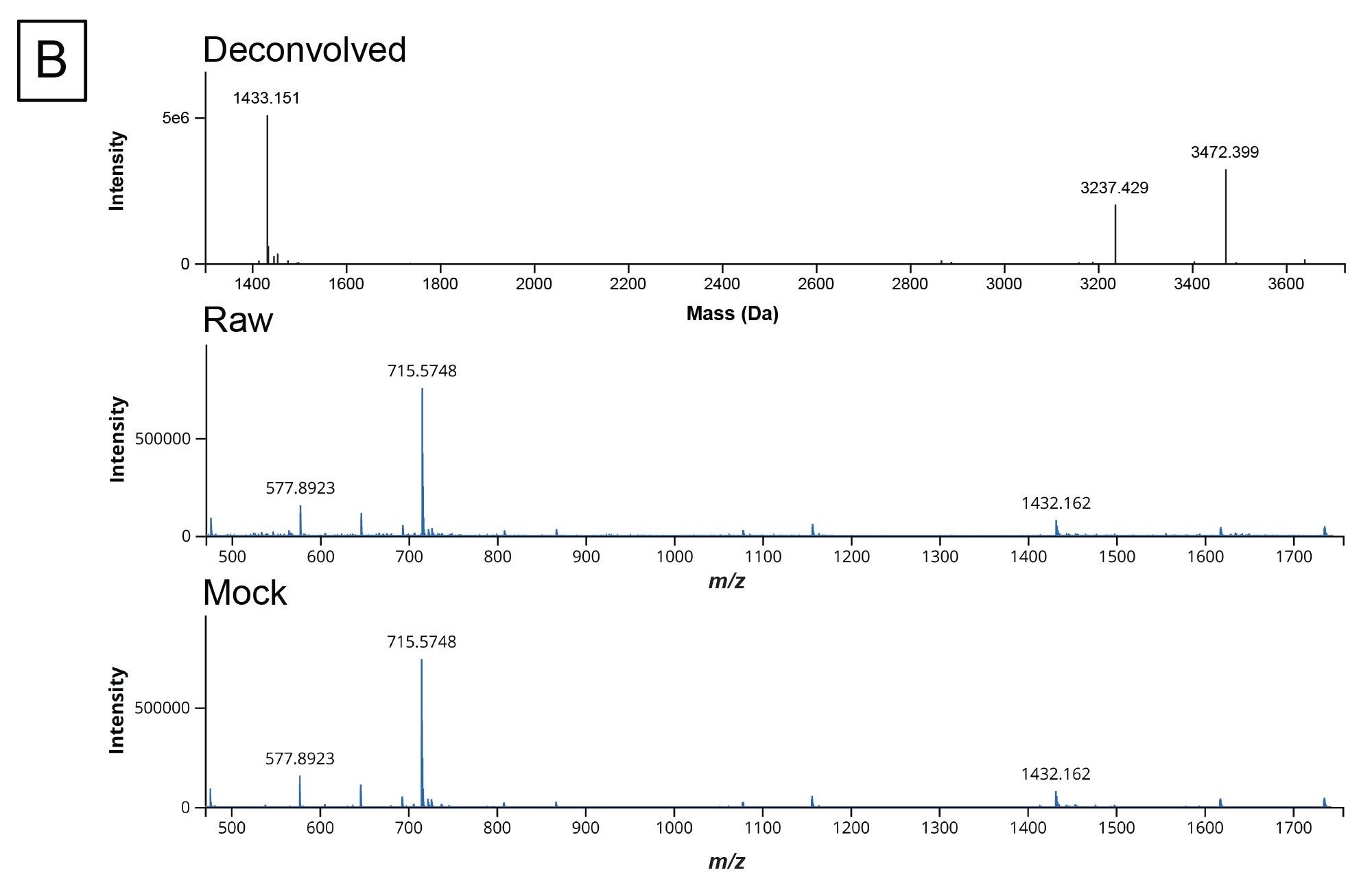 Figure 4. (B) An example of the deconvoluted spectra output.
Figure 4. (B) An example of the deconvoluted spectra output.
While the tabular data produced by the INTACT Mass App is the key result for reporting fragment mapping data, the Coverage Viewer microApp can be used for visualization of these results against the sequence, and calculation of sequence coverage. The GUI of the Coverage Viewer microApp is featured in Figure 5. The sequence of the RNA molecule of interest is entered into the sequence field and the mRNA Cleaver output generated in the first step of the workflow can be imported to populate the target digest fragment list (bottom left-hand corner). The INTACT Mass results file can be imported using the “Import” button on the left-hand side. The sequence coverage is visualized on the right-hand side of the GUI and the coverage percentage is calculated as targets are confirmed. If the INTACT Mass results file is imported, digestion fragments that have the keyword “Pass” will be considered “Confirmed”. Manually assigned products can also be checked off by the user to be considered “Confirmed” as well. The red colored area under the relevant sequence will change to green to indicate that specific part of the sequence has been confirmed by accurate mass measurement. Once finished, the user can then export the target digested fragment section with the “Confirmed” column as a .csv for later use. Additionally, the coverage map image can be copied by clicking “Capture image to clipboard” and saved in another program.
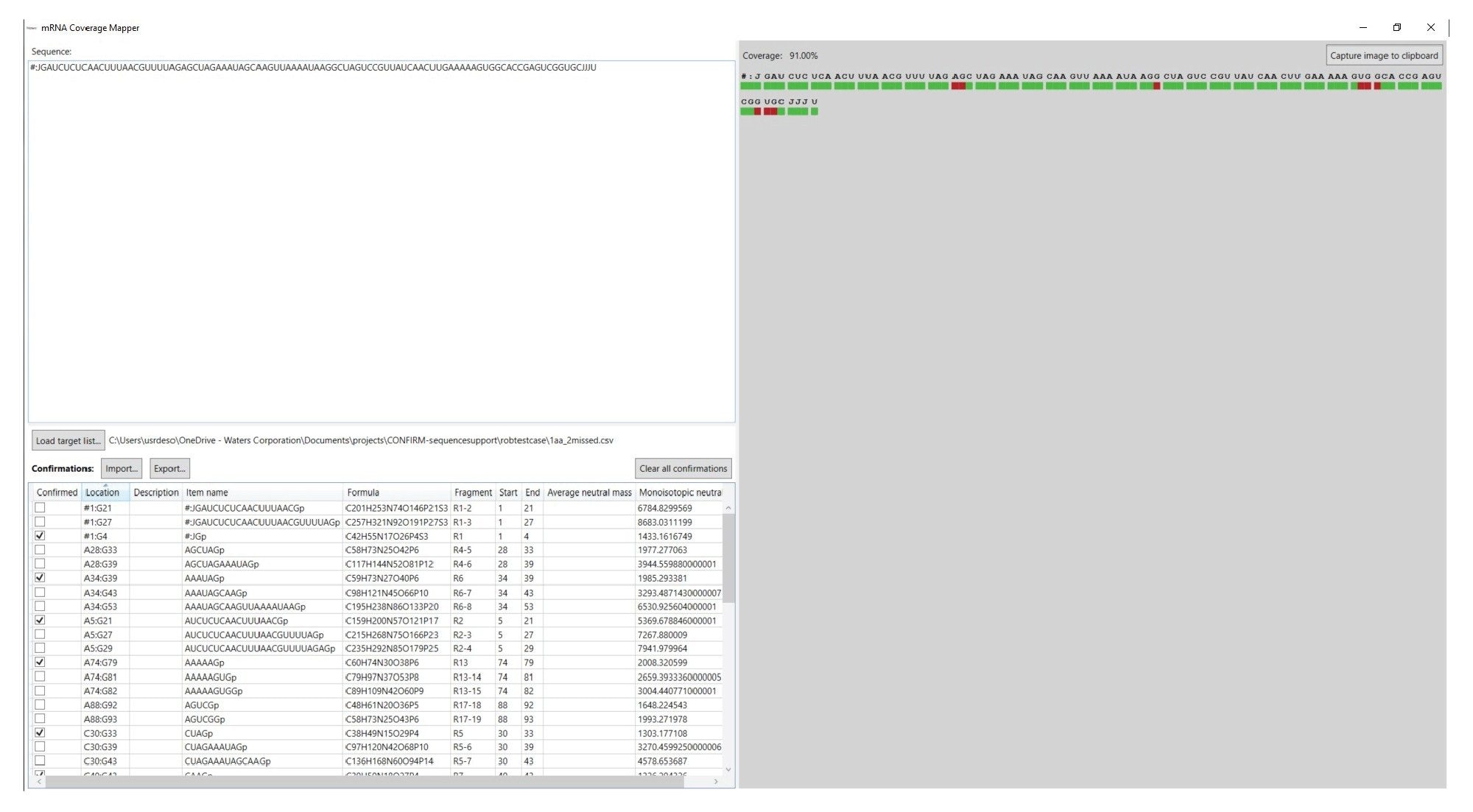 Figure 5. The Coverage Viewer microApp User Interface.
Figure 5. The Coverage Viewer microApp User Interface.
3. Test case: sgRNA digestion and mapping
The CRISPR-based gene editing process has become popular in recent years, largely due to its simplicity.16 The process uses two main components, guide RNA and the CRISPR-associated (Cas) nuclease.16 The guide RNA is a specific RNA sequence that recognizes the target DNA of interest and directs the Cas nuclease to the correct site for editing .16 The Cas nuclease is a non-specific nuclease which depends entirely on the guide RNA to act upon a specific DNA locus.16 Originally, guide RNA was composed of two distinct oligonucleotides, the Crispr RNA, which is complementary to the target, and the tracr RNA, which served as a binding scaffold for the Cas nuclease.16 As the technology evolved the crispr RNA and the tracr RNA were fused together with a region called the “linker loop” and became single guide RNA or sgRNA.16 The key CQA for synthetically generated sgRNA is confirming its sequence, including the modified residues engineered into specific sites on the molecule.
By performing a RNase T1 digestion and analyzing the digestion products using our workflow, we can identify the digestion products and confirm, at the MS1 level (without additional MS fragmentation), the map coverage of the sgRNA sequence. First, the sgRNA modified nucleotides were added to the residues.csv file to allow mRNA Cleaver to correctly determine the in silico digestion products of the sgRNA. Then, we adjusted the digestion and MS settings of the mRNA Cleaver microApp to reflect the experimental conditions, specifically that RNase T1 was the enzyme used and that we would like mRNA Cleaver to generate digestion fragments with two missed cleavages. Even in a complete digestion, there is still a chance the enzyme will not cleave at every G and by increasing the missed cleavage variable to two, we account for that possibility. The in silico digestion product list was then generated and exported into a .csv file. Two ambiguous sequences, 4mer oligos with the sequence CUAG, were identified. Ambiguous sequences are digestion fragments that are isomeric and/or isobaric, which means the m/z peaks and charge state envelopes for these sequences will overlap. Ambiguous sequences that cover a portion of the RNA that is critical to analysis may require further experimental steps to confirm sequence identity such as fragmentation in the gas phase. Otherwise, the other 19 digestion fragments have unique expected masses and sequence identities, which should be discernable in our mass spectra. We base this assumption upon the expected mass accuracy generated by the BioAccord, and the in silico generated m/z range for each digestion fragment from the mRNA Cleaver microApp.
The in vitro digestion was performed as described in the experimental methods and the data for both the digested sgRNA and the non-digested intact sgRNA was collected on the BioAccord LC-MS system using waters_connect. Figure 6A depicts the total ion chromatograms for the intact sgRNA and the subsequent digestion. The digested fragments eluted between five and 40 minutes of the gradient. The sgRNA has been completely digested as the intact sgRNA elutes at 35.6 minutes and there is no corresponding peak in the digestion sample trace.
The LC-MS data and the list of in silico digestion components were both submitted to the INTACT Mass App to assign the various digestion components based on a mass match with a 10 ppm match tolerance. We then imported the INTACT Mass results into the Coverage Viewer microApp to visualize the sequence coverage, shown in Figure 6B. We were able to achieve 91% MS1 sequence coverage. For this test case, we did not perform collision induced fragmentation to ascertain sequence identity as our ambiguous sequences were the same sequence identity so it would be superfluous. However, one may want to exclude ambiguous sequences from calculating sequence coverage when there are many ambiguous sequences or ambiguous sequences of interest that have differing sequence identities unless the sequence identity of these ambiguous sequences can be confirmed by fragmentation.28 To achieve 100% sequence coverage, one could use another endoribonuclease such as MasF or RNase A that have differing cleavage specificity to RNase T1 to cover the unidentified portion of the sgRNA sequence.
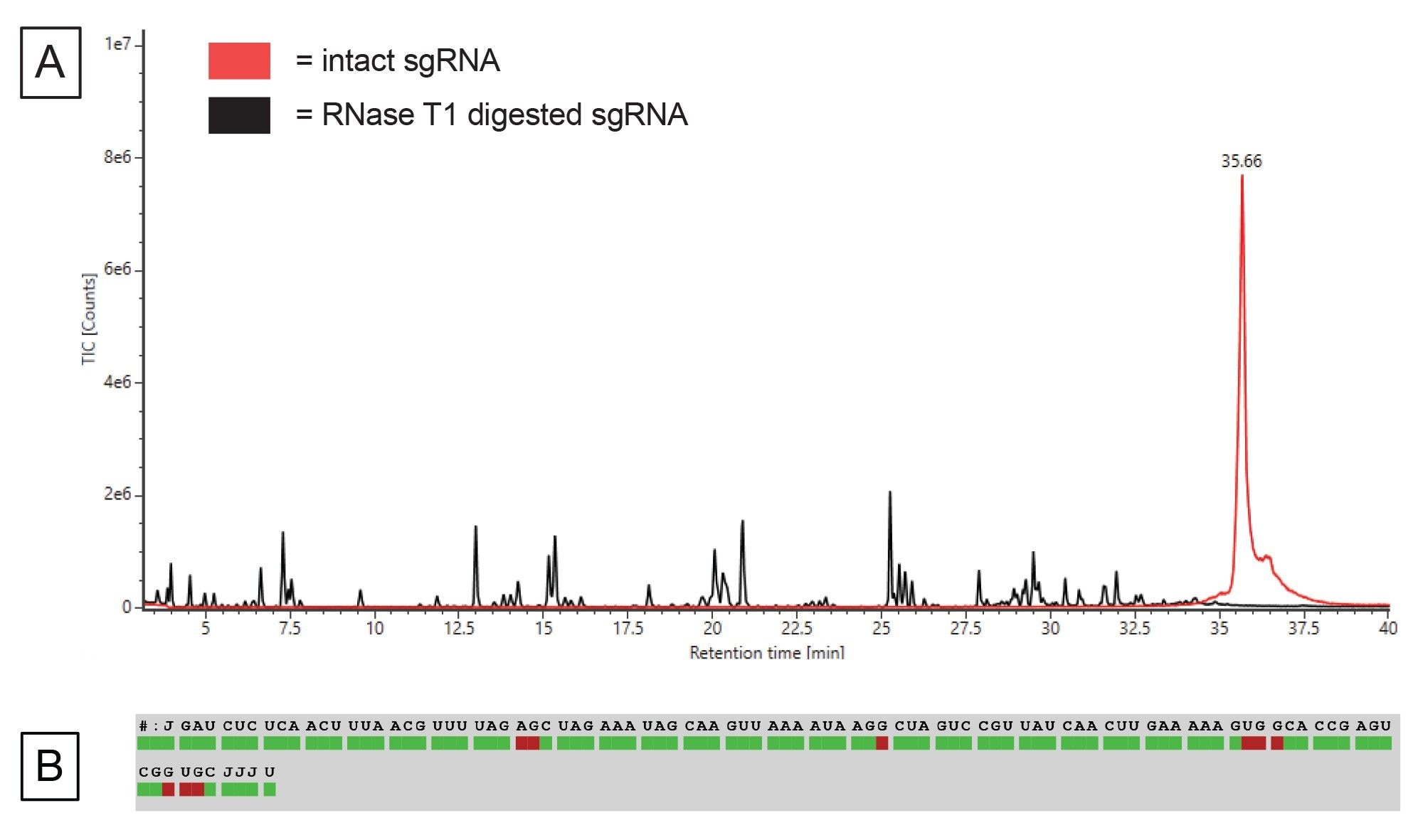 Figure 6. (A) TIC of sgRNA digested (black trace) and the undigested intact sgRNA (red trace), (B) Sequence coverage afforded by LC-MS via the Coverage Viewer microApp.
Figure 6. (A) TIC of sgRNA digested (black trace) and the undigested intact sgRNA (red trace), (B) Sequence coverage afforded by LC-MS via the Coverage Viewer microApp.
mRNA digestion and mapping
This workflow can be applied to larger RNAs, such as messenger RNA (mRNA). However, mapping larger RNAs at the MS1 level is ambitious as there will be many additional ambiguous sequences and coeluting species, exponentially increasing the data complexity. Gaye et al has previously released an application note regarding methodology to optimally perform mRNA sequence mapping using IP-RP LC-MS and the waters_connect platform.18 Here, we have reprocessed the data using the updated workflow.
Figure 7A contains the TIC chromatogram from the RNase T1 mRNA digest from Gaye et al as well as the Coverage Viewer results.18 The inset shows the extracted mass spectrum(XIC) from the peak eluting at 8.58 minutes. This representative XIC highlights the complexity of the data as multiple species are coeluting under the peak. Following the workflow, the mRNA Cleaver microApp was used to generate two in silico enzymatic digestion product lists: one including the ambiguous sequences and one excluding them. These list and the acquired LC-MS data were submitted to the INTACT Mass App for data processing. The results from INTACT Mass were then imported into the Coverage Viewer microApp.
The results from INTACT Mass were then imported into the Coverage Viewer microApp. The MS1 level digestion fragment maps indicated 38.87% unique sequence coverage and 75.18% overall sequence coverage (ambiguous sequences included). These maps are illustrated in Figure 7B and 7C respectively. While this sequence coverage can be improved using another endoribonuclease in parallel, the complexity of the data (coeluting species, ambiguous sequences, etc.) will continue to be a challenge for data interpretation. Data processing time for these more complex samples scales with the number of peaks analyzed and can become limiting to analysis throughput.
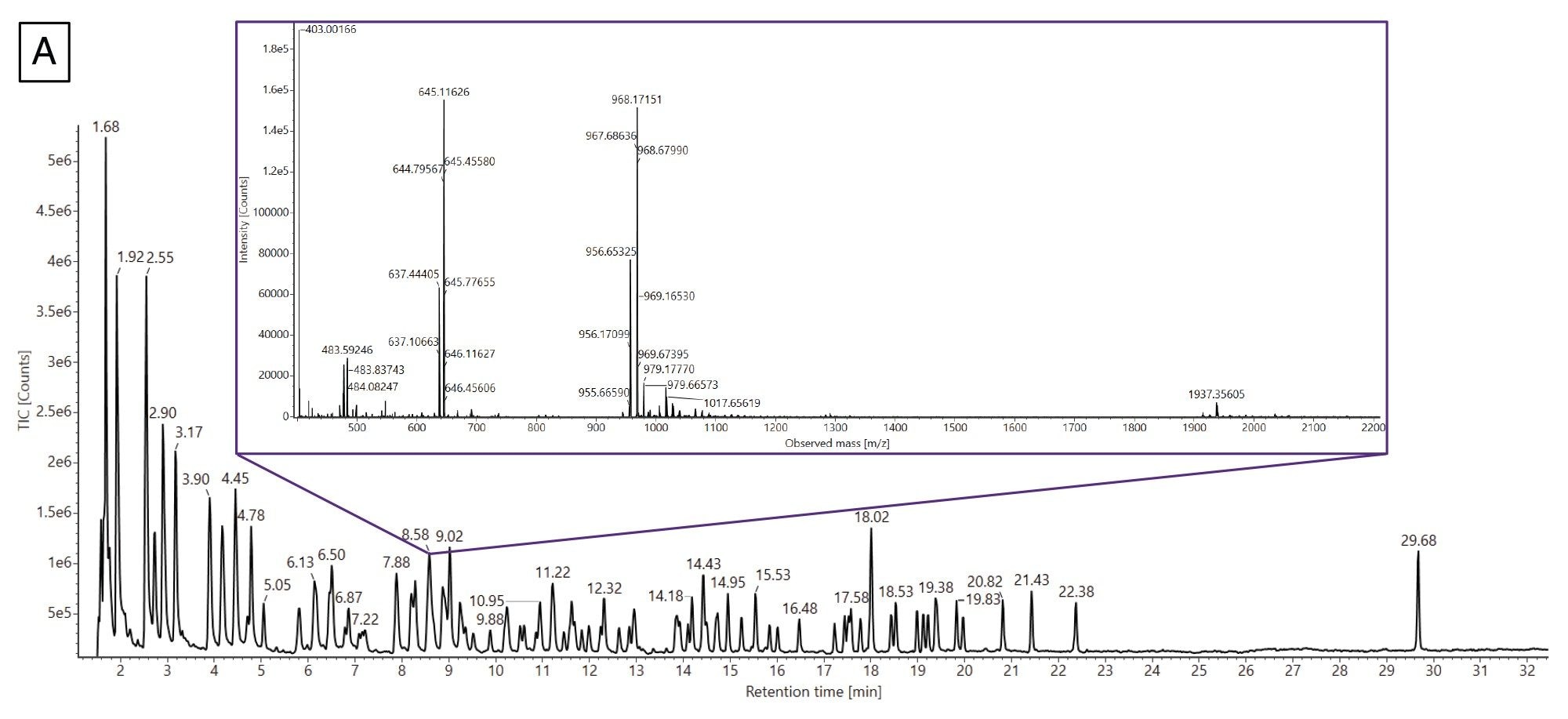 Figure 7. (A) TIC of the mRNA digest from Gaye et al.(18) The inset spectrum is a representative XIC from the peak eluting at 8.58 min that contains several oligonucleotide species that are coeluting.
Figure 7. (A) TIC of the mRNA digest from Gaye et al.(18) The inset spectrum is a representative XIC from the peak eluting at 8.58 min that contains several oligonucleotide species that are coeluting.
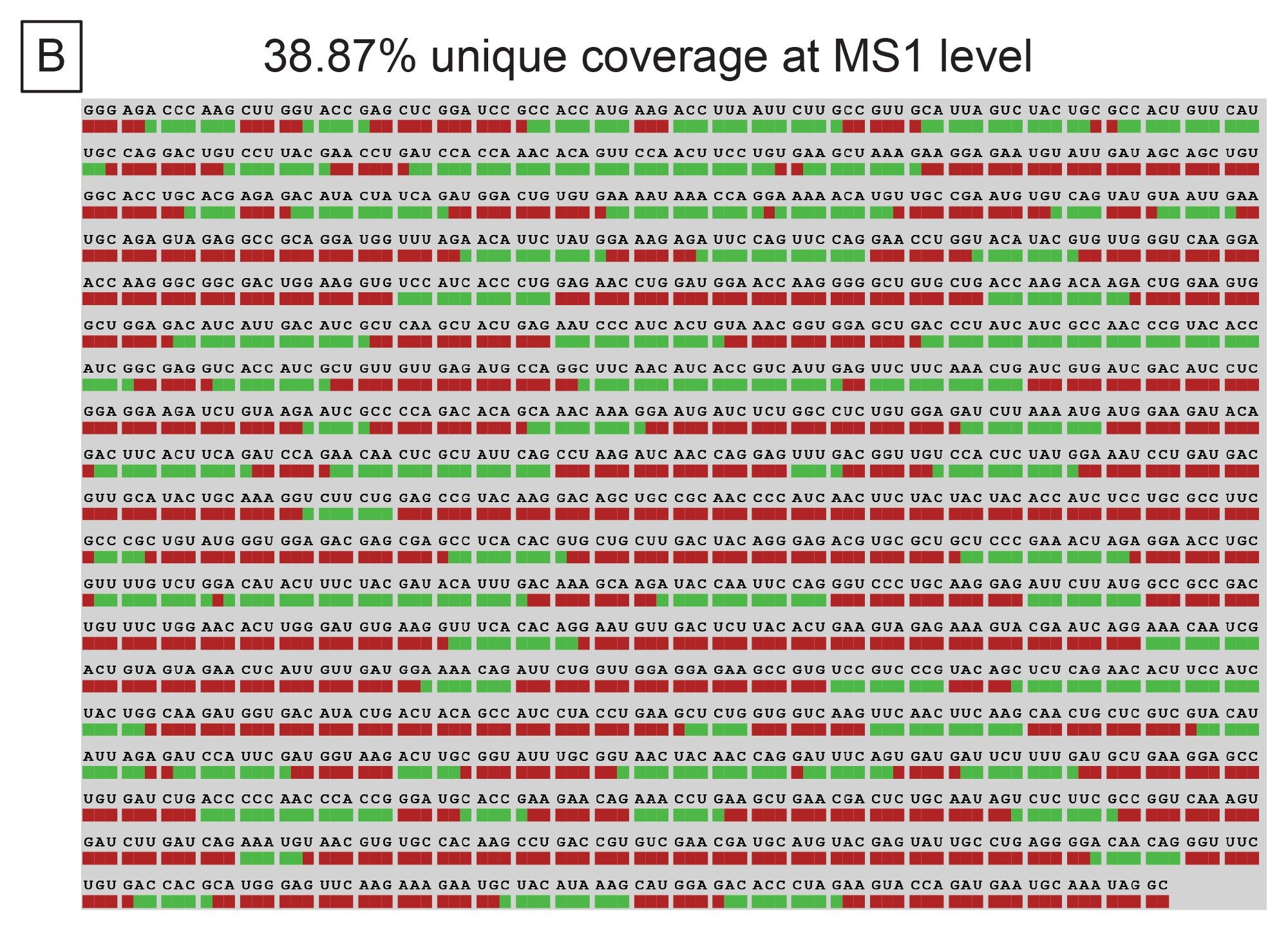 Figure 7. (B) Unique sequence coverage (no ambiguous sequences included) afforded by MS1 via the Coverage Viewer microApp.
Figure 7. (B) Unique sequence coverage (no ambiguous sequences included) afforded by MS1 via the Coverage Viewer microApp.
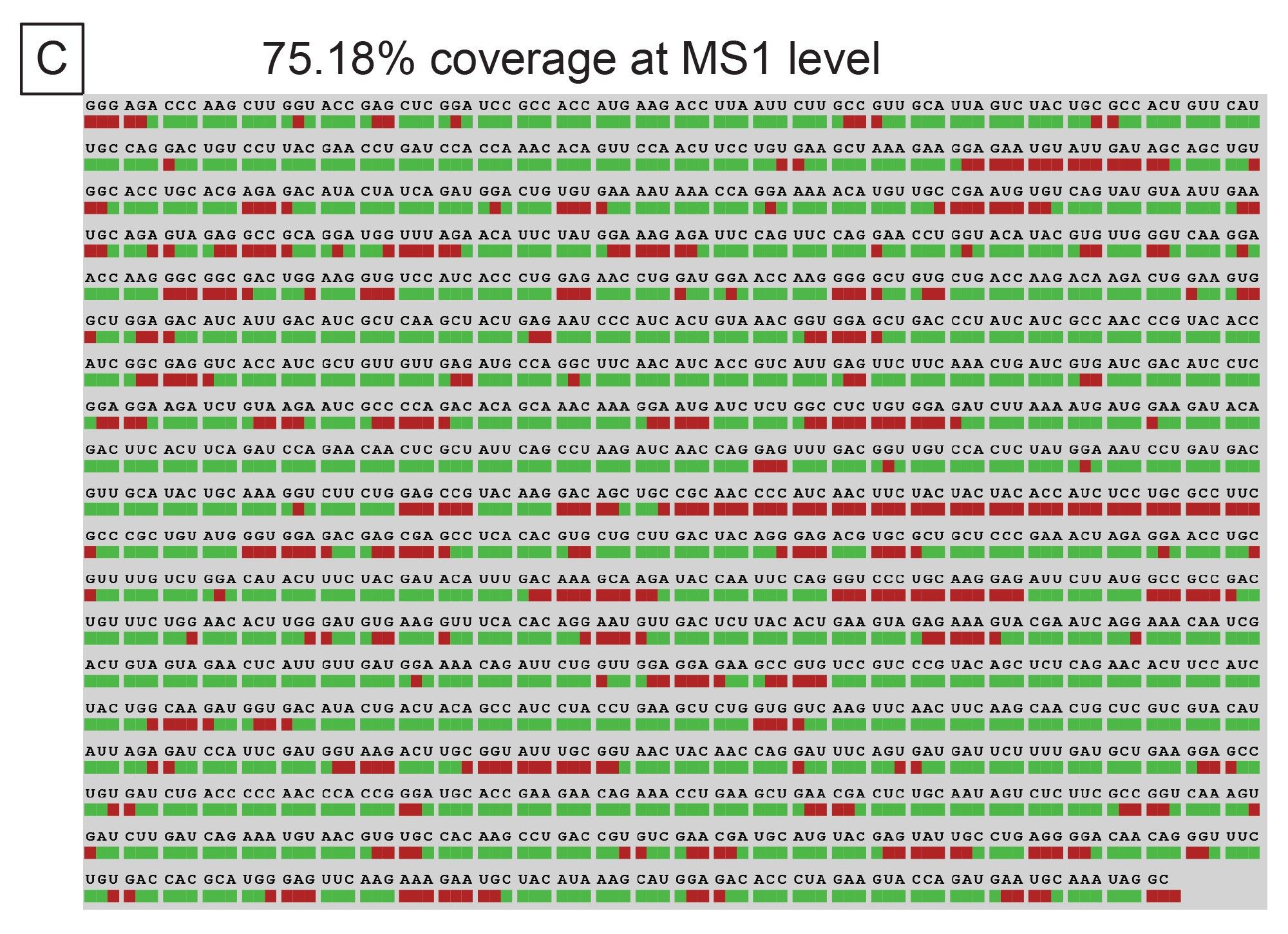 Figure 7. (C) Overall sequence coverage afforded by MS1 via the Coverage Viewer microApp.
Figure 7. (C) Overall sequence coverage afforded by MS1 via the Coverage Viewer microApp.
Further sequence elucidation of ambiguous fragments can be done using the CONFIRM Sequence App if data independent (MSE) fragmentation data is collected as part of the mapping experiment on the BioAccord. An example of this type of analysis from Gaye et al is shown in Figure 8.18 Workflow automation of this additional stage of analysis is ongoing.
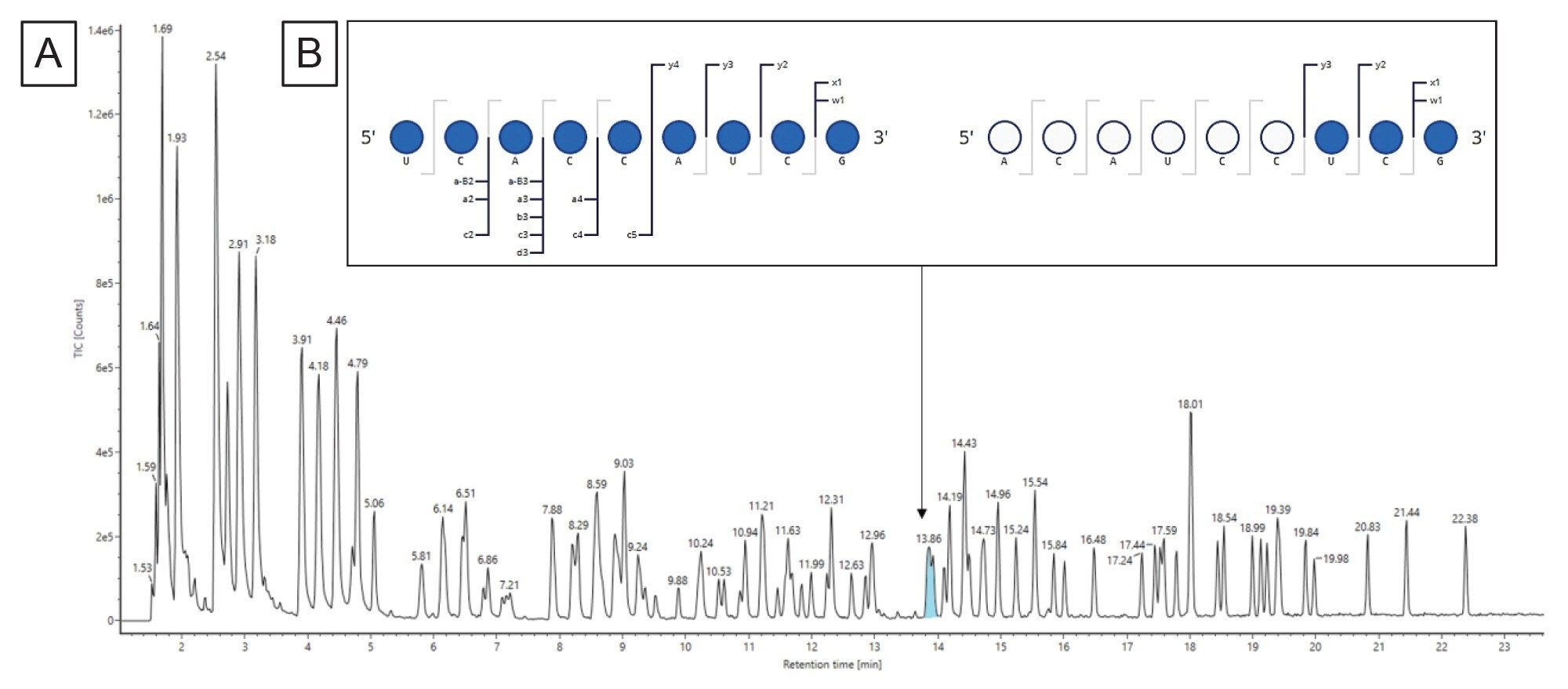 Figure 8. (A) Digested fragment components at position 623–631(ACAUCCUCGp) and 551–559 (UCACCAUCGp) are predicted and assigned to the same RT peak in the TIC from Gaye et al. It is not possible to determine the correct assignment using intact mass information. (B) MSE data from the same injection can be used to elucidate the correct sequence for this assignment. Using the waters_connect CONFIRM Sequence application, high energy fragment ions are predicted for each sequence and matched to isotope clusters of the integrated raw data via a bespoke algorithm. The software presents confirmed fragment ions on a Dot-Map to quickly assess the sequence coverage.
Figure 8. (A) Digested fragment components at position 623–631(ACAUCCUCGp) and 551–559 (UCACCAUCGp) are predicted and assigned to the same RT peak in the TIC from Gaye et al. It is not possible to determine the correct assignment using intact mass information. (B) MSE data from the same injection can be used to elucidate the correct sequence for this assignment. Using the waters_connect CONFIRM Sequence application, high energy fragment ions are predicted for each sequence and matched to isotope clusters of the integrated raw data via a bespoke algorithm. The software presents confirmed fragment ions on a Dot-Map to quickly assess the sequence coverage.
Measuring 5’ capping effectiveness
A CQA specific to mRNA is to assess the 5’ capping efficiency and structure. The 5’ cap of a mRNA molecule is essential for several biological functions including: regulation of nuclear export, prevention of degradation by exonucleases, promotion of translation, and promotion of 5’ proximal intron excision.14 In a previous application note, Nguyen et al. described a method for the rapid analysis (under five minutes) of synthetic mRNA cap structure with the BioAccord LC-MS System.14
Using our workflow, we were able to detect and assign the 5’ cap impurities based on the deconvoluted spectra as shown in Figure 9A. To analyze this data, we applied custom charge deconvolution, a new feature of the INTACT Mass App (as of release version 1.6.0). Auto deconvolution uses optimized settings for peak detection and deconvolution processing and is appropriate for simplifying routine analysis of many samples of varying levels of complexity. However, this data had a large dynamic range of analytes, including several low intensity impurities near lower detection limits of the analysis that we wanted to robustly identify, and we found that the auto deconvolution algorithm settings were not appropriate in this case. Using custom deconvolution allows scientists to optimize the spectral deconvolution settings and better represent the full population of 5’ cap structures present on the molecule.
For this data, we applied the BayesSpray charge deconvolution algorithm and tailored the retention time range, the input m/z range, the output expected mass range, and charge range to our data. The INTACT Mass data for the 5’ cap analysis is shown in Figure 9B. We were able to identify all of the impurities (ppG, pppG, GpppG, and m7GpppG (Cap-0)) in the 1:10 dilution sample and three of the aforementioned impurities (excluding pppG) in the 1:100 dilution sample from Ngyuen et al.14
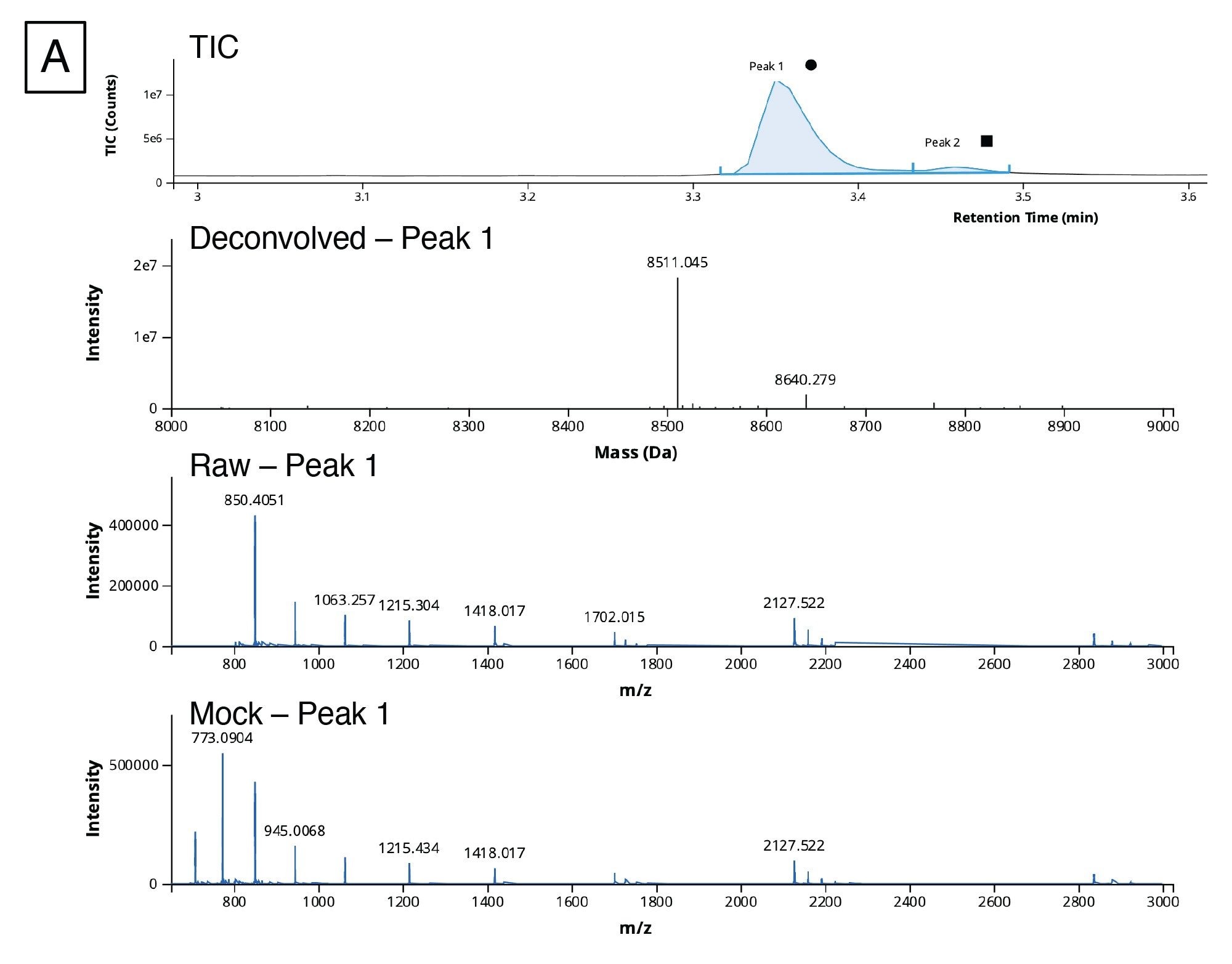 Figure 9. (A) TIC, Deconvoluted, Raw, and Mock spectra for the peak at 3.37 minutes (circle).
Figure 9. (A) TIC, Deconvoluted, Raw, and Mock spectra for the peak at 3.37 minutes (circle).
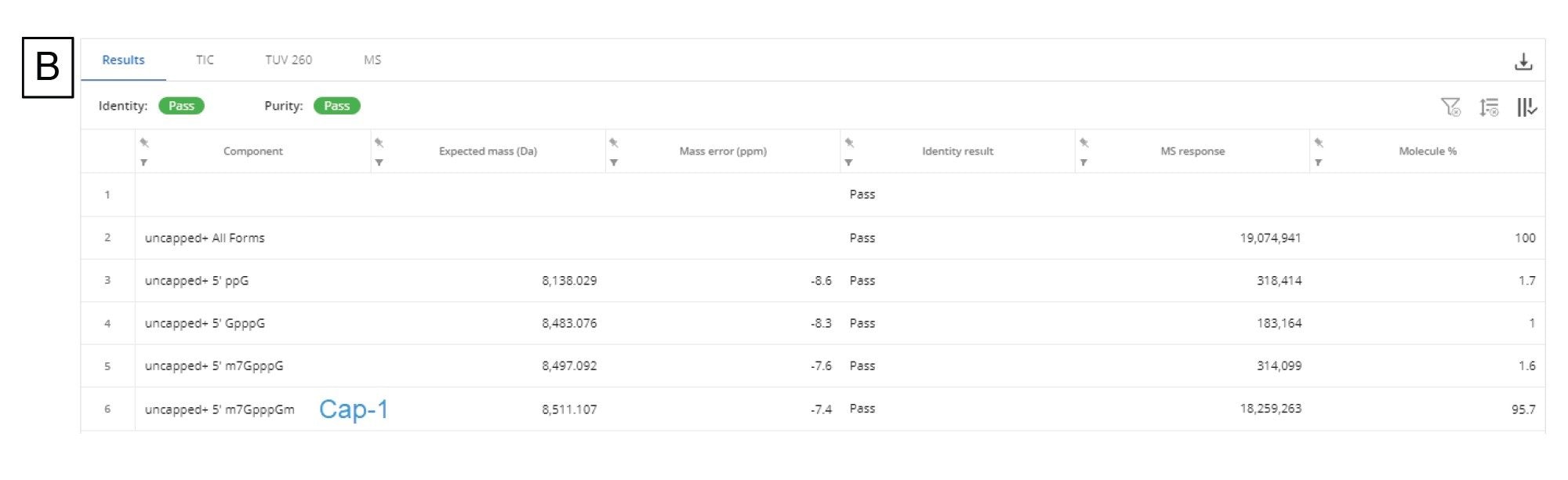 Figure 9. (B) INTACT Mass App results table for the Cap-1 fragment and its product related impurity fragments.
Figure 9. (B) INTACT Mass App results table for the Cap-1 fragment and its product related impurity fragments.
Effective translation is critically dependent upon the 5’ cap of mRNA and therefore the percentage of correctly capped mRNA product is a closely monitored CQA in the manufacturing of mRNA therapeutic products. The assay developed by Nguyen et al and the data analysis workflow using custom deconvolution settings is sensitive and robust enough to routinely identify unwanted impurities arising from an imperfect capping process.14
Achieving reliable 3’ poly(A) tail length and distribution measurements
Another CQA specific to mRNA is to determine the 3’ poly(A) tail length. The 3’ poly(A) tail length is directly correlated with mRNA stability.17 Therefore, determining the length, or rather the average length and size distribution, can impart valuable insight into the polyadenylation process. A relatively fast (15 min runtime) LC-MS assay was developed by Doneanu et al for the analysis of 3’ poly(A) Tail oligonucleotides produced from Firefly Luciferase (FLuc) mRNA, following RNase T1 digestion.13 As shown in Figure 10A, the 3’ poly(A) Tail elutes as a well resolved, abundant, late-eluting chromatographic peak in the TIC. The corresponding combined ESI-MS spectrum, containing a population of coeluting 3’ poly(A) species, was submitted to the workflow and deconvolved using the MaxEnt1 charge deconvolution algorithm available in the INTACT Mass App. Custom charge deconvolution parameters, such as the input m/z range, the output mass range, and retention time range, were adjusted to produce the deconvolved spectrum shown in the inset of Figure 10A. This spectrum indicates a broad 3’ poly(A) heterogeneity, with oligonucleotides ranging in size from 122 to 132 mers differing by the characteristic 329.2059 Da adenosine monophosphate residue mass. The INTACT Mass results are shown in Figure 10B and the ESI-MS signals and their corresponding intensities of all detected oligo components are summarized in a plot displayed in Figure 10C. This graph was used to calculate the weight-averaged mass of the 3’ poly(A) Tail of the FLuc mRNA. The measured average mass corresponds to the length of a 126.5 mer, and this measurement was confirmed from orthogonal SEC measurements.15 Following INTACT Mass processing, the eleven 3’ poly(A) variants were measured with typical mass accuracies better than 45 ppm (Figure 10B).
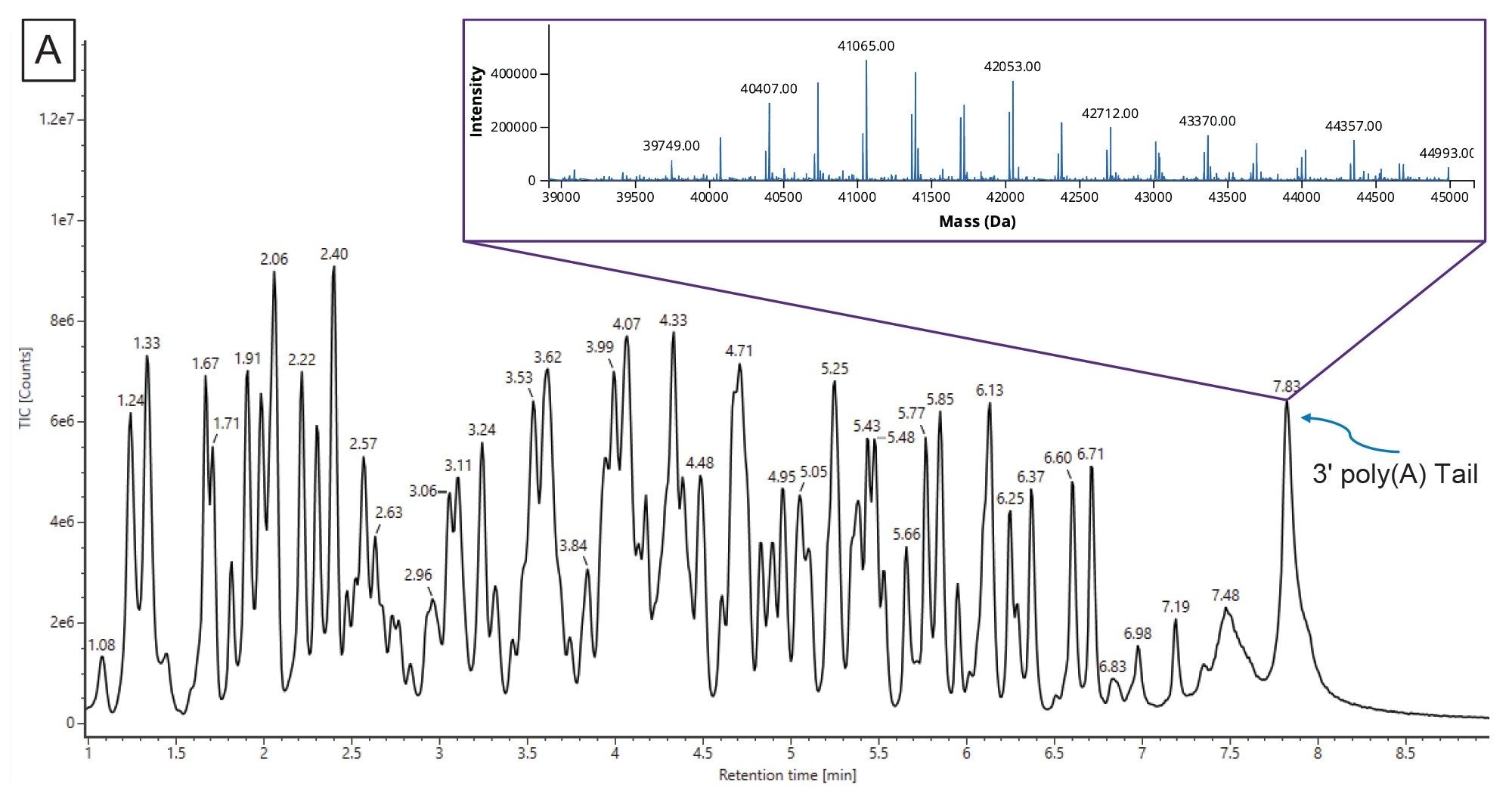 Figure 10. (A) TIC chromatogram of the 3’ poly (A) Tail oligonucleotide mixture derived from RNase T1 digested FLuc mRNA. The inset illustrates the MaxEnt1 charge deconvolution from the INTACT Mass Application.
Figure 10. (A) TIC chromatogram of the 3’ poly (A) Tail oligonucleotide mixture derived from RNase T1 digested FLuc mRNA. The inset illustrates the MaxEnt1 charge deconvolution from the INTACT Mass Application.
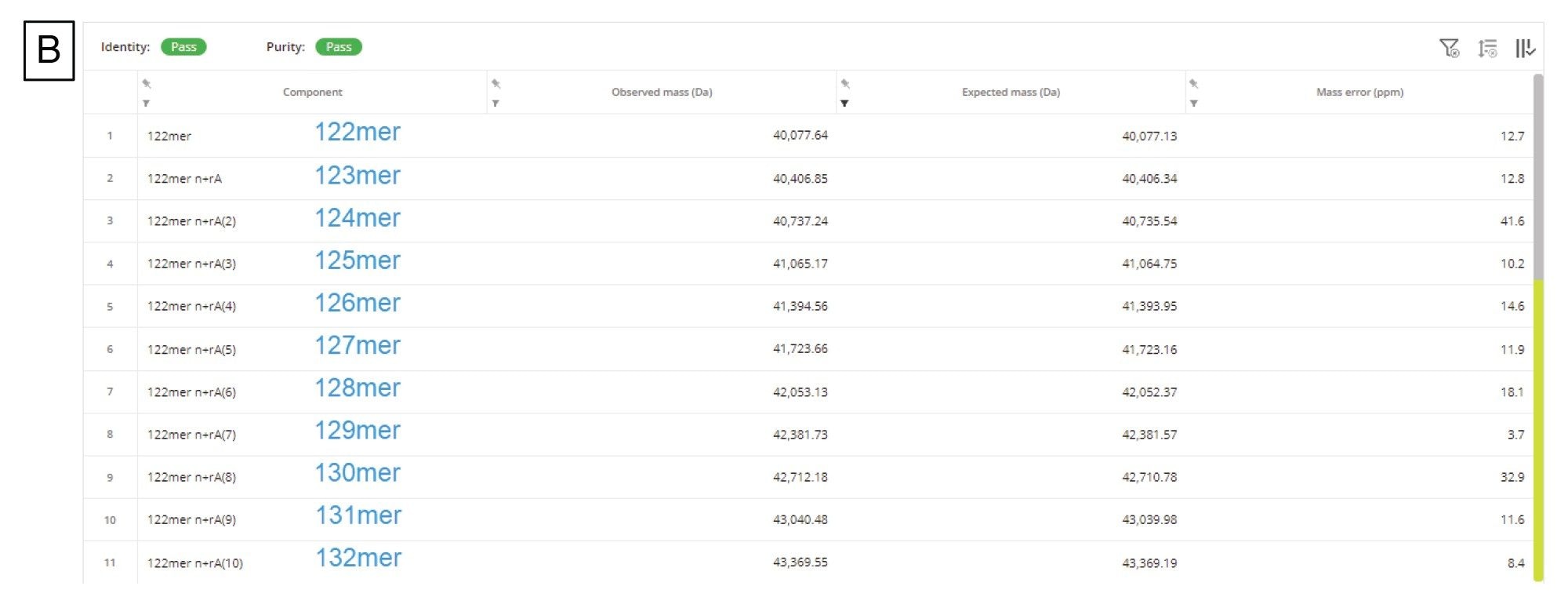 Figure 10. (B) The INTACT Mass Application results table illustrating the wide dispersity of the 3’ poly(A) tail feature, with up to eleven adenosine masses added to the first detected tail variant, a 122 -mer. Eleven oligonucleotide species, ranging from 122- to 132-mers were identified with mass accuracies better than 45 ppm using INTACT Mass processing with custom deconvolution parameters.
Figure 10. (B) The INTACT Mass Application results table illustrating the wide dispersity of the 3’ poly(A) tail feature, with up to eleven adenosine masses added to the first detected tail variant, a 122 -mer. Eleven oligonucleotide species, ranging from 122- to 132-mers were identified with mass accuracies better than 45 ppm using INTACT Mass processing with custom deconvolution parameters.
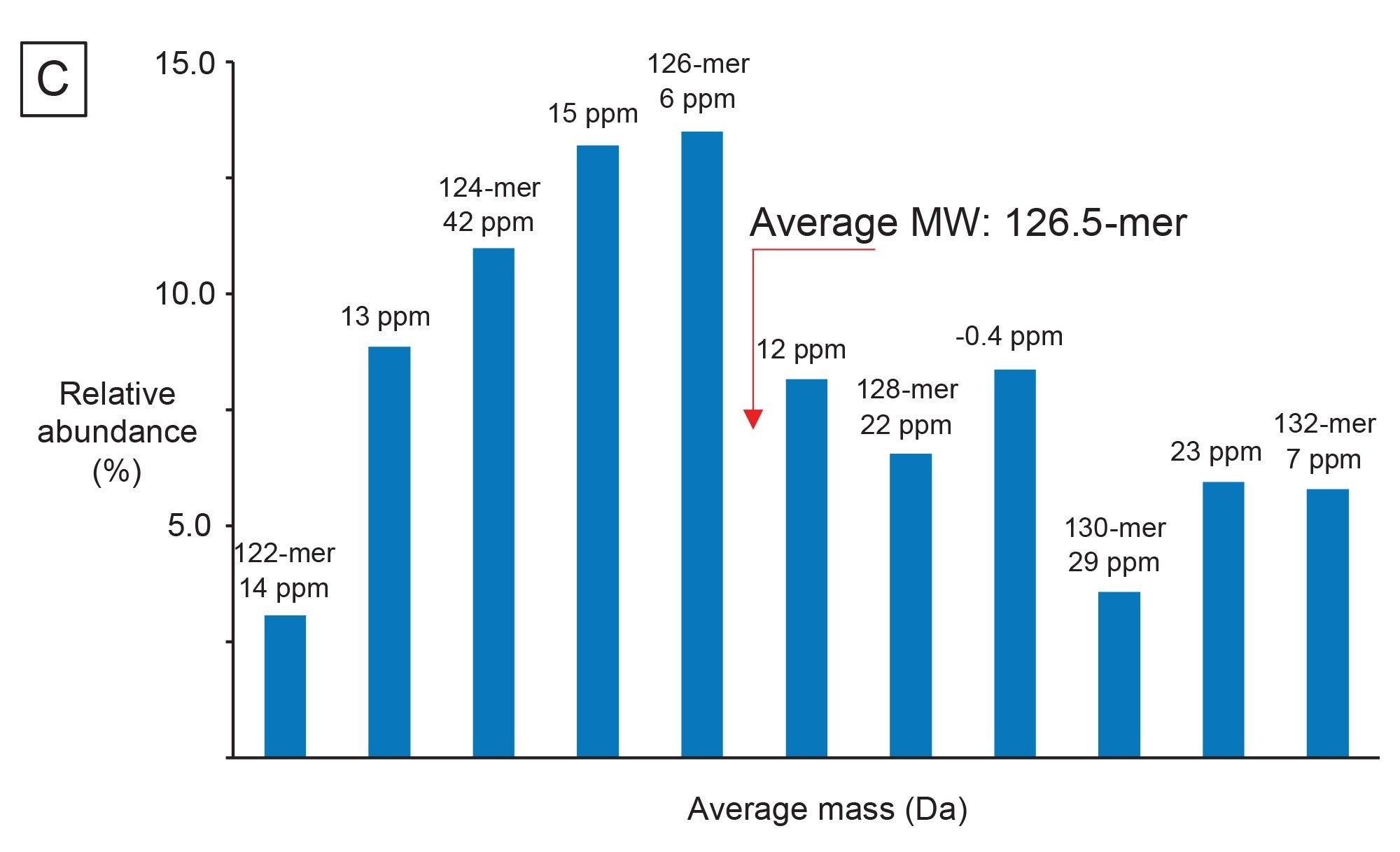 Figure 10. (C) The distribution of the ESI-MS spectral intensity versus the 3’ poly(A) tail length enables average tail length and dispersity to be determined.
Figure 10. (C) The distribution of the ESI-MS spectral intensity versus the 3’ poly(A) tail length enables average tail length and dispersity to be determined.
Both the 3’ poly(A) dispersity as well as the average mass measurement are important CQAs for therapeutic mRNA molecules, measuring process robustness, and potential clinical effect. Thus, the LC-MS assay developed by Doneanu et al and our data analysis workflow using the INTACT Mass App proves effective for analysis of 3’ poly(A) Tails enzymatically cleaved from prophylactic/therapeutic mRNAs.
Conclusion
- CQAs for longer oligos and modern RNA therapeutics can be analyzed by approaches including sequence map confirmation (oligos, sgRNA, and mRNA), 5’ capping effectiveness (mRNA), and 3’ poly(A) tail analysis (mRNA) via workflows built on the BioAccord LC-MS System and the waters_connect informatics platform.
- Informatics enhancements to the INTACT Mass application were produced to support these workflows. The new custom deconvolution capability enables routine automated data processing, even with samples of low signal intensity and high dynamic range for the analytes of interest.
- These enhancements to the INTACT Mass application are complemented by additional microApps developed for RNA data analysis:
- mRNA Cleaver that generates an in silico digestion list with user editable residues, enzymes, and mRNA modifiers
- Coverage Viewer that allows for easy visualization and calculation of digestion fragment map sequence coverage
References
- Zhu, Y., Zhu, L., Wang, X. & Jin, H. RNA-based Therapeutics: An overview and prospectus. Cell Death & Disease 13, (2022).
- Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA-Targeted Therapeutics. Cell Metab. 2018 Apr 3;27(4):714-739. doi: 10.1016/j.cmet.2018.03.004. Erratum in: Cell Metab. 2019 Feb 5;29(2):501. PMID: 2961764
- Damase, T. R. et al. The Limitless Future of RNA therapeutics. Frontiers in Bioengineering and Biotechnology 9, (2021).
- Xu, S.; Yang, K.; Li, R.; Zhang, L., mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. International Journal of Molecular Sciences 2020, 21 (18), 6582
- Verbeke, R., Lentacker, I., De Smedt, S. C. &; Dewitte, H. The dawn of mRNA vaccines: The COVID-19 case. Journal of Controlled Release 333, 511–520 (2021).
- Jackson, N. A., Kester, K. E., Casimiro, D., Gurunathan, S. &; DeRosa, F. The Promise of mRNA vaccines: A biotech and industrial perspective. npj Vaccines 5, (2020).
- Goyon, A. et al. Full sequencing of CRISPR/Cas9 single guide RNA (sgRNA) via parallel ribonuclease digestions and hydrophilic interaction liquid chromatography–high-resolution mass spectrometry analysis. Analytical Chemistry 93, 14792–14801 (2021).
- Ivleva, V. B., Yu, Y.-Q. &; Gilar, M. Ultra-performance liquid chromatography/tandem mass spectrometry (UPLC/MS/MS) and UPLC/MSe analysis of RNA oligonucleotides. Rapid Communications in Mass Spectrometry 24, 2631–2640 (2010).
- Jiang, T. et al. Oligonucleotide sequence mapping of large therapeutic mRNAs via parallel ribonuclease digestions and LC-MS/MS. Analytical Chemistry 91, 8500–8506 (2019).
- Ross, R., Cao, X. & Limbach, P. Mapping post‐transcriptional modifications onto transfer ribonucleic acid sequences by liquid chromatography tandem mass spectrometry. Biomolecules 7, 21 (2017).
- Shion, H., Boyce, P., Berger, S. & Yu, Y. Q. INTACT Mass™ - A Versatile waters_connect™ application for Rapid Mass Confirmation and Purity Assessment of Biotherapeutics. Waters Application Note, 720007547, February 2023.
- Jackson, N. A., Kester, K. E., Casimiro, D., Gurunathan, S. & DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. npj Vaccines 5, (2020).
- Doneanu, C., Fredette, J. & Yu, Y. Q. Ion-Pairing Reversed Phase LC-MS Analysis of Poly(A) Heterogeneity Using the BioAccord LC-MS System, Waters Application Note, 720007925, 2023.
- Nguyen, J. M. et al. Rapid analysis of synthetic mRNA cap structure using ion-pairing RPLC with the BioAccord LC-MS System. Waters Application Note, 720007329, October 2023.
- Gilar, M. Size-exclusion chromatography method for poly(A) tail analysis of mRNA. Waters Application Note, 720007853, October 2023.
- Hu, L., Li, Y., Wang, J., Zhao, Y. & Wang, Y. Controlling CRISPR-Cas9 by guide RNA engineering. WIREs RNA 14, (2022).
- Pelletier, J.; Sonenberg, N. The Organizing Principles of Eukaryotic Ribosome Recruitment. Annu. Rev. Biochem. 2019, 88, 307–335
- Gaye, M. M. et al. Synthetic mRNA oligo-mapping using ion-pairing liquid chromatography and mass spectrometry. Waters Application Note, 720007669, October 2023.
- Fekete, S et al. Challenges and emerging trends in liquid chromatography-based analyses of mRNA pharmaceuticals. Journal of Pharmaceutical and Biomedical Analysis. 224 (2022).
- Fountain, K. J., Gilar, M. & Gebler, J. C. Analysis of native and chemically modifed oligonucleotides by tandem ion-pair reversed phase high-performance liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 17, 646–653. https://doi.org/10.1002/rcm.959 (2003).
- Zhang, G., Lin, J., Srinivasan, K., Kavetskaia, O. & Duncan, J. N. Strategies for bioanalysis of an oligonucleotide class macromolecule from rat plasma using liquid chromatography−tandem mass spectrometry. Anal. Chem. 79, 3416–3424. https://doi.org/10.1021/ac0618674 (2007).
- Deng, P., Chen, X., Zhang, G. & Zhong, D. Bioanalysis of an oligonucleotide and its metabolites by liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. Anal. 52, 571–579. https://doi.org/10.1016/j.jpba.2010.01.040 (2010).
- Gau et al. Oligonucleotide mapping via mass spectrometry to enable comprehensive primary structure characterization of an mRNA vaccine against SARS‑CoV‑2. Nature Scientific Reports. 13:9038 2023.
- Loverix S, Winqvist A, Stromber R, Steyaert J. Mechanism of RNase T1: concerted triester-like phosphoryl transfer via a catalytic three-centered hydrogen bond Chem Biol. 2000 Aug, 7(8):651-8 doi: 10.1016/s1074-5521(00)00005-3.
- Loverix S, Steyaert J. Deciphering the Mechanism of RNase T1. Methods in Enzymology, Academic Press, Vol. 341, 2001, 305-323, doi: 10.1016/S0076-6879(01)41160-8
- Masachika Irie. 3- RNase T1/RNase T2 Family RNases. Ribonucleases, Academic Press, 1997. 101-130, doi: 10.1016/B978-012588945-2/50004-2.
- Sorrentino, Salvatore. The Eight Human “Canonical” Ribonucleases: Molecular Diversity, catalytic properties, and special biological actions of the enzyme proteins. FEBS Letters. Vol 584, Issue 11, 2010, 2194-2200, doi: 10.1016/j.febslet.2010.04.018
- Gaye M.M. et al. CRISPR Single Guide RNA Characterization by IP-RP-LC-MS with a Premier Oligonucleotide BEH 300Å C18 Column. Waters Application Note, 720007897, October 2023.
720008130, November 2023