People involved in polymer characterization by chromatographic techniques do not exclusively use GPC to analyze their samples. Many times we need to use liquid chromatographic techniques by adsorption or partition chromatography to get the information we need.
Conventional reverse-phase and, at times, normal phase separation techniques are used to quantitate polymer additives, as an example. Obtaining the molecular weight distribution of your polymer sample may be just one part of the characterization process. What about the additives that are formulated into the polymer to offer stabilization or processing enhancement? They can be even more important than the polymer itself. We need to think about using the correct UV stabilizers and antioxidants for protection against degradation, plasticizers to improve flexibility, antistats for polyolefins, flame retardants, accelerators to enhance the crosslinking (or curing) process, and so forth.
We have done an extensive amount of work with polymer additives, and you can find some of our published work detailed in the Journal of Liquid Chromatography, volume 14 #3, (1991) and volume 16, #7, (1993).
How do we analyze polymer additives? First, we need to think of what we are trying to accomplish. Do we need to know if the correct amounts of each additive are present in the formulation? Are we trying to "deformulate" a competitive material? Do we need to extract the additive package out from the polymer matrix? Chances are, the answers to these questions will be "Yes". GPC analysis is not the best way to separate, identify and quantitate the levels of additives present. Most of the additives are quite close to each other in size and molecular weight, so we need to use HPLC to separate them. A simple gradient technique, with optional flow programming, works very well in getting many different types of additives separated in a short run time. A gradient analysis consists of varying the eluent, or mobile phase composition, usually from a "weak" solvent to a "strong" solvent over a period of time. This composition variation is usually done in a linear fashion for additive analysis. Since we are varying the eluent composition throughout the chromatographic run, the refractive index detector can not be used.
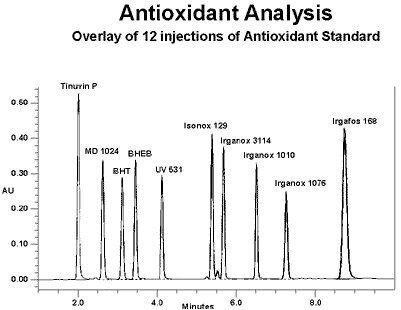
Most of the polymer additives we deal with have some chromophore that absorbs ultraviolet light, so a UV detector is used primarily. If there are no chromophores present, an evaporative light scattering detector may be used. We can also change the flow rate throughout the run, usually increasing flow to get the later eluters to come out more quickly. The column usually chosen for additive analysis is either an octadecylsilane (C18) or Octylsilane (C8) column, ~15 cm in length. An example of a reverse-phase gradient (with flow program) separation of a series of common antioxidants and UV stabilizers 9 overlay of 12 injections is shown here.



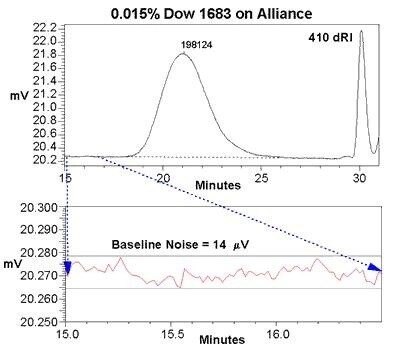
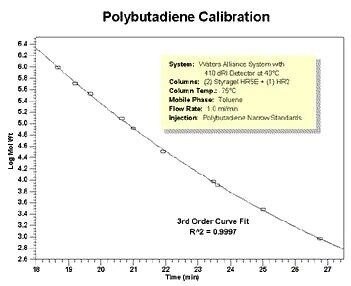




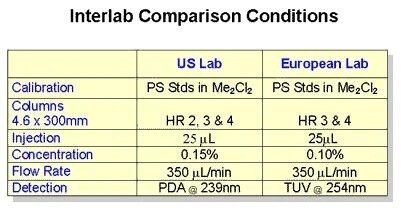








.jpg.82.resize/img.jpg)