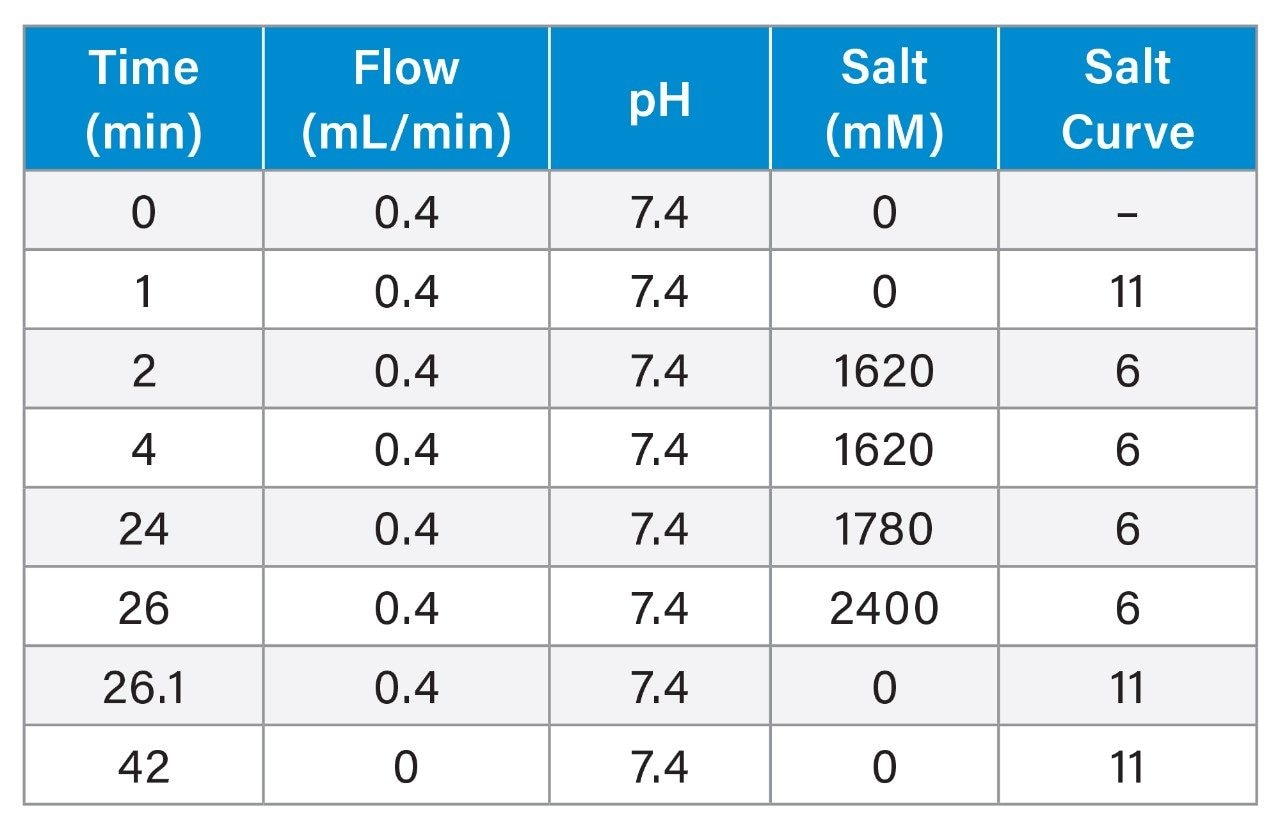
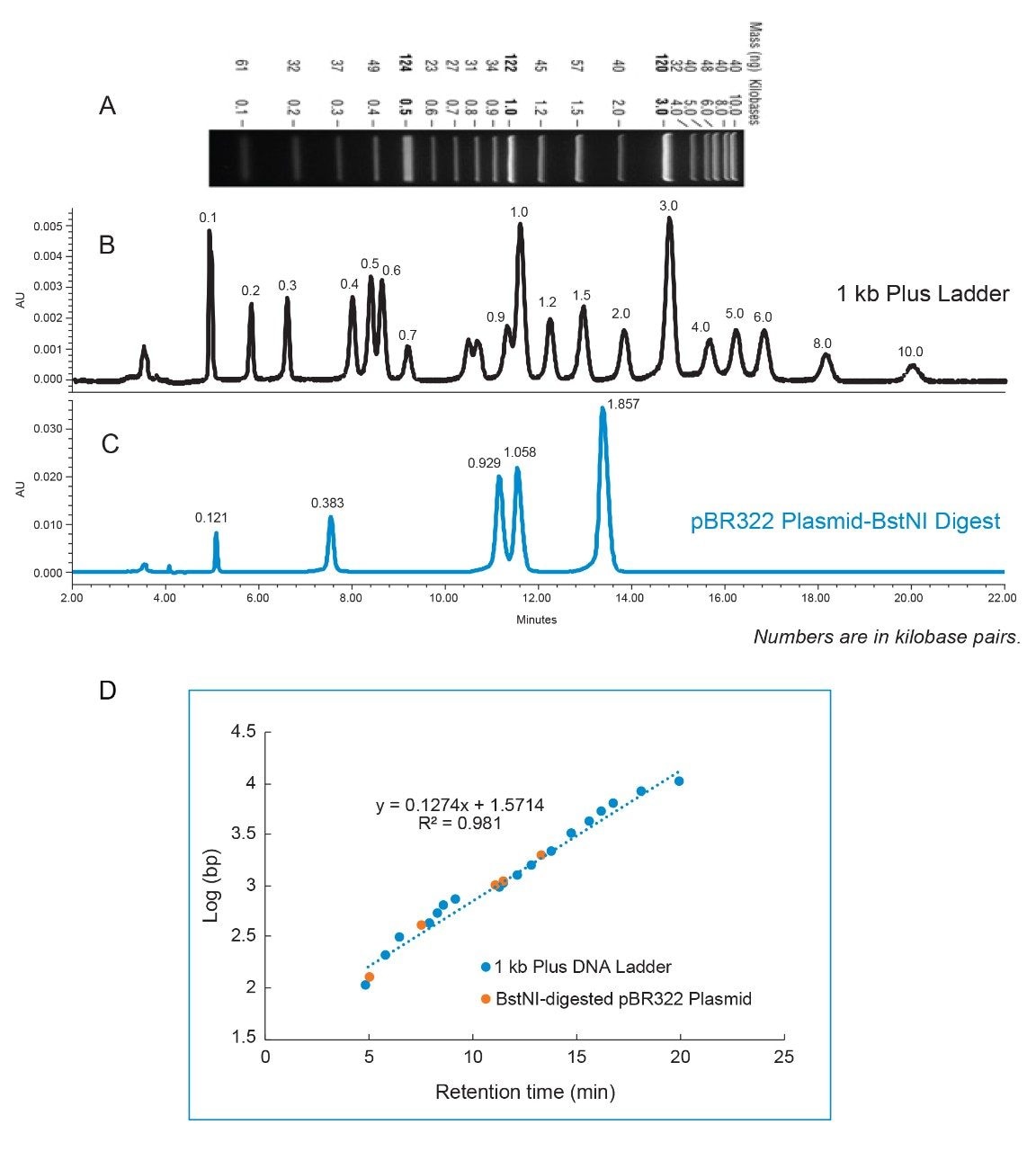
As shown in Figure 1B, 1 kb Plus DNA Ladder that consists of a series of dsDNA fragments ranging from 0.1 kbp to 10 kbp was separated on a Waters Protein-Pak Hi Res Q Strong Anion-Exchange Column. The ability of separating a wide range of different sizes of dsDNA is essential for the analysis of a restriction enzyme digestion.
The chromatographic separation is consistent with the agarose gel separation provided on the New England Biolabs website (Figure 1A and 1B). To help identify the size of the fragments easily, the amount of 1 kb and 3 kb fragments were intentionally made to be higher than other fragments in the 1 kb Plus dsDNA Ladder, which can be observed on the gel with the 1 kbp and the 3 kbp bands being thicker and brighter. Consistently, the peak areas of the 1 kbp and the 3 kbp fragments are also higher than those of other fragments on the anion-exchange chromatogram.
Since the number of negatively charged phosphodiester groups is proportional to the number of nucleotides, the DNA fragments should elute according to their size. Generally, this is true when the DNA fragments are short (<100 bp). As the DNA size increases, the composition of the DNA may have some impact on the AEX separation. For example, it has been shown that fragments having high A-T content elute later than expected based on their chain lengths.2,3 In some cases, the shorter fragments having higher A-T content eluted later than the longer fragments having lower A-T content.4 This can be problematic for size assessment since the DNA does not elute in the order of its length. In these attempts, NaCl was used as the eluting salt in AEX separation. Interestingly, it has been shown that the melting temperature of the A-T rich DNA is higher than that of the G-C rich DNA in NaCl. The difference between A-T rich DNA and G-C rich DNA in the melting temperature is abolished when the experiments were carried out in tetramethylammonium chloride (TMAC). It has been hypothesized that TMAC can fit into the grooves at DNA structure and preferentially bind to A-T base pair, resulting in composition-independent melting temperature for DNAs.5,6
In our previous work, we showed that TMAC provides better separation power than NaCl as an eluting salt for analytes involving nucleic acid.7,8 In the current study, TMAC is again experimented as the salt to separate various length of dsDNA fragments. Preliminary data from Multi-Angle Light Scattering (MALS) suggest that the molecular weights of the peaks in the chromatogram increases along with the retention time (data not shown). Although it is not completely clear why TMAC is able to separate DNA fragments based on their size independent of the fragment composition, it is assumed that TMAC plays an important role in eliminating the influence of the compositions in the dsDNA fragments on the final AEX separation outcome.
Traditionally, the size of the DNA fragments is assessed by running the fragment DNA on the agarose gel and comparing the position of the bands with that of the DNA Ladder that serves as a reference material run on the same gel under the same conditions. Similarly, we show here that in an AEX method, retention time can be used to assess the size of the DNA fragment by running it with the 1 kb Plus DNA Ladder under the same chromatographic conditions (Figure 1B and 1C). Figure 1D shows a plot of log(bp) versus retention time of the dsDNA fragments. The blue dots are the data points from the 1 kb Plus DNA Ladder, while the orange dots are those from the BstNI-digested pBR322 plasmid. The linear fit from the 1 kb Plus DNA Ladder indicates a strong correlation between the logarithm of the size and the retention time (R2 = 0.981). Using this plot, the sizes of the restriction fragments are calculated from their retention times. The percent (%) error is calculated using the formula: {(calculated size – theoretical size)/theoretical size}. The measurement for most restriction fragments from pBR322 plasmid resulted in <11% error, which is highlighted by the orange dots that are located on or very close to the trendline in the plot. The % error for the first restriction fragment (0.121 kbp) is greater than that of the other DNA fragments. This deviation is likely attributed to the data point for the early eluting fragment in the 1 kb Plus DNA Ladder, 0.1 kbp, which also resides far from the trendline.