Sample description
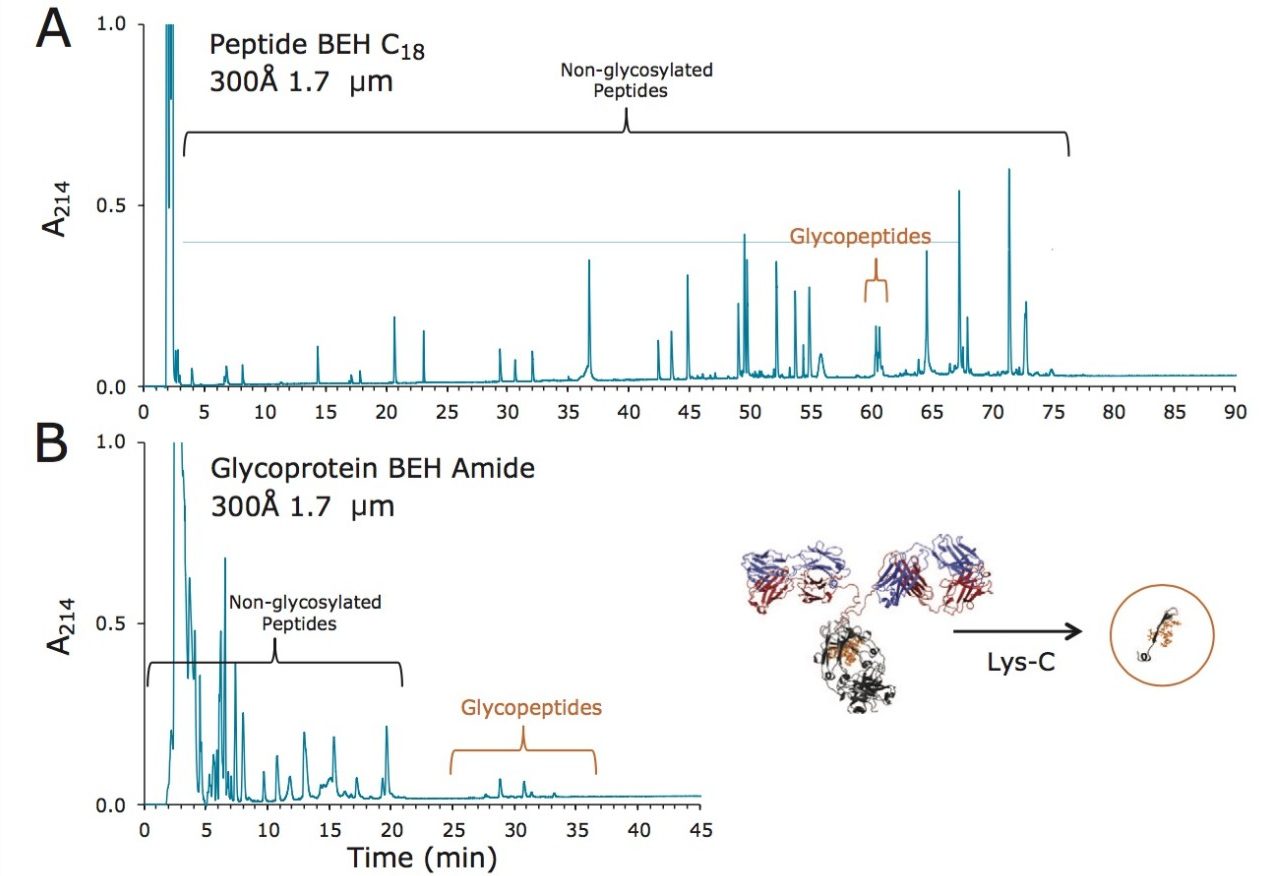
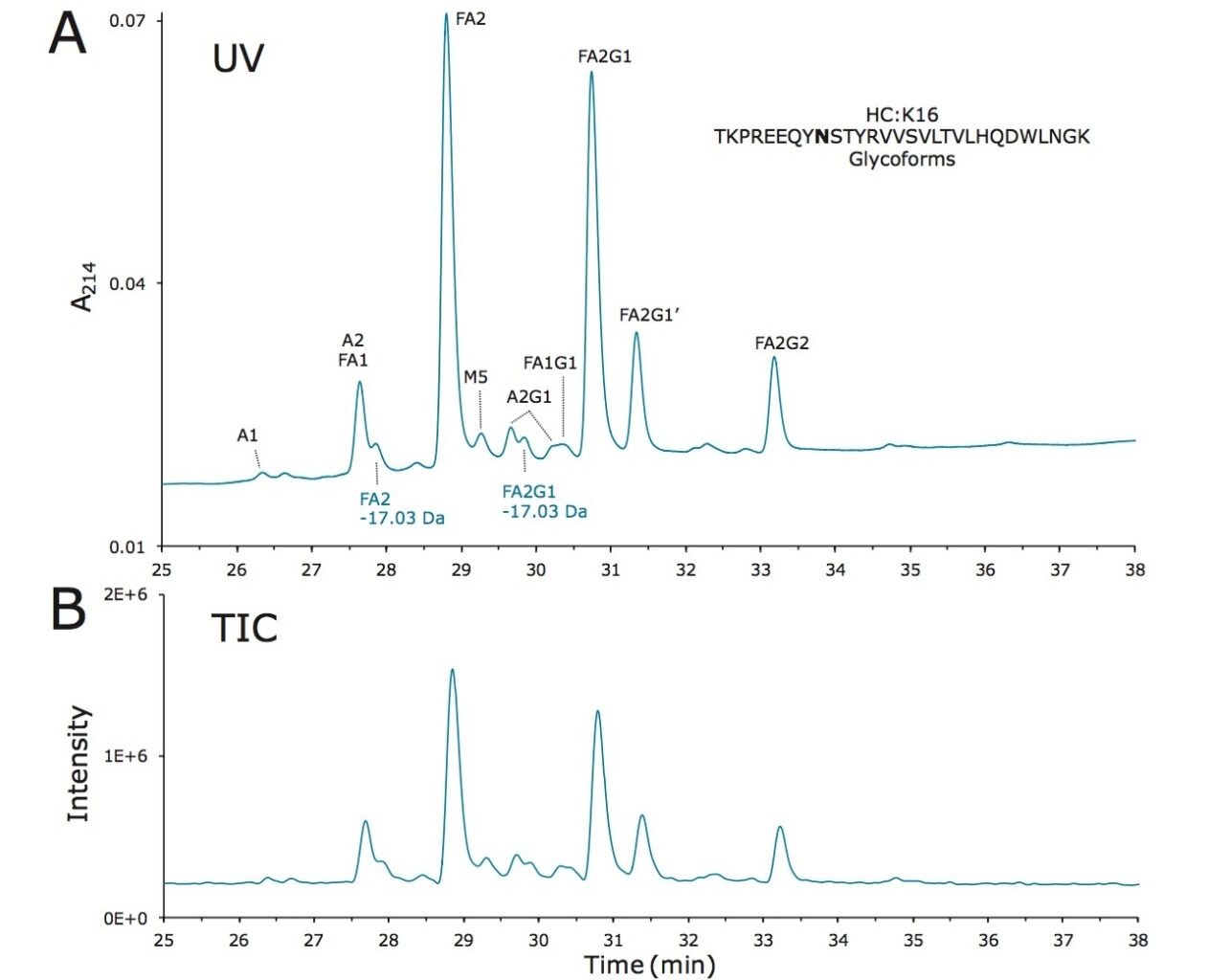
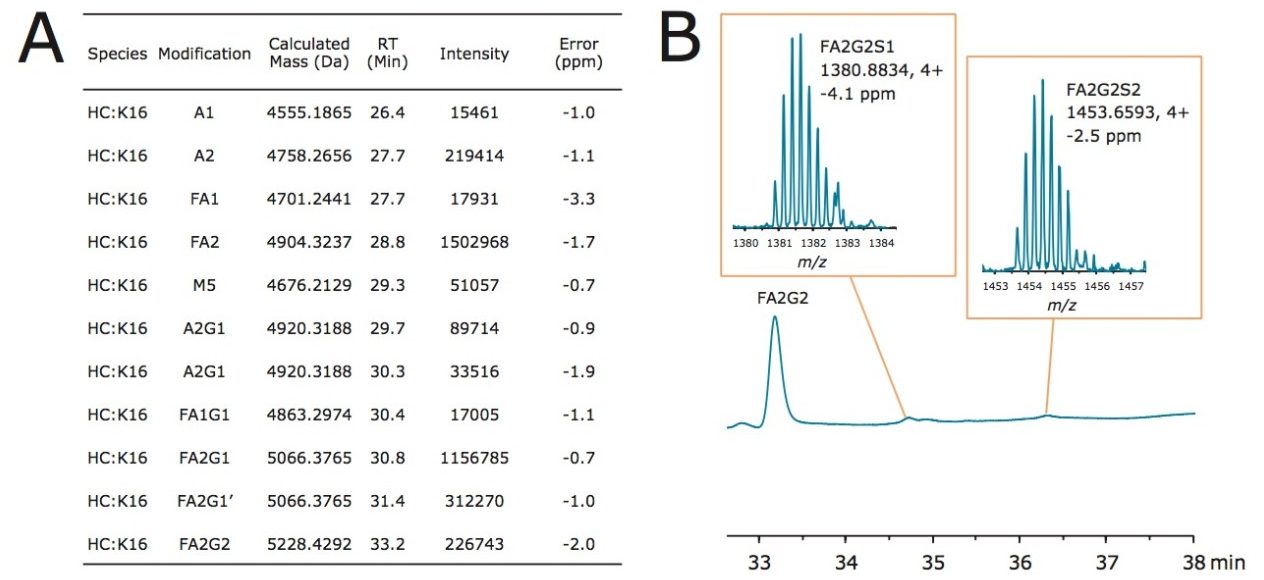
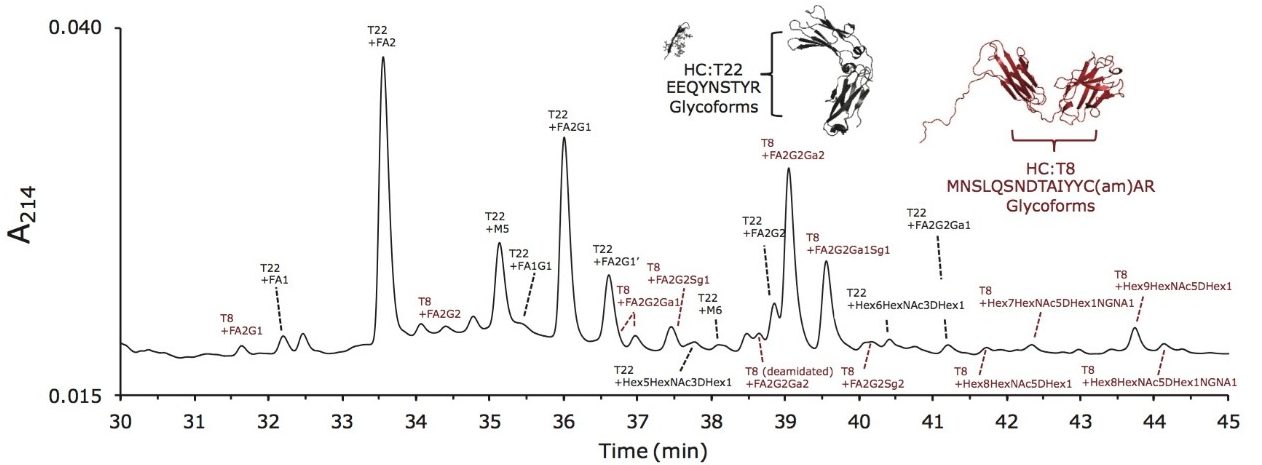
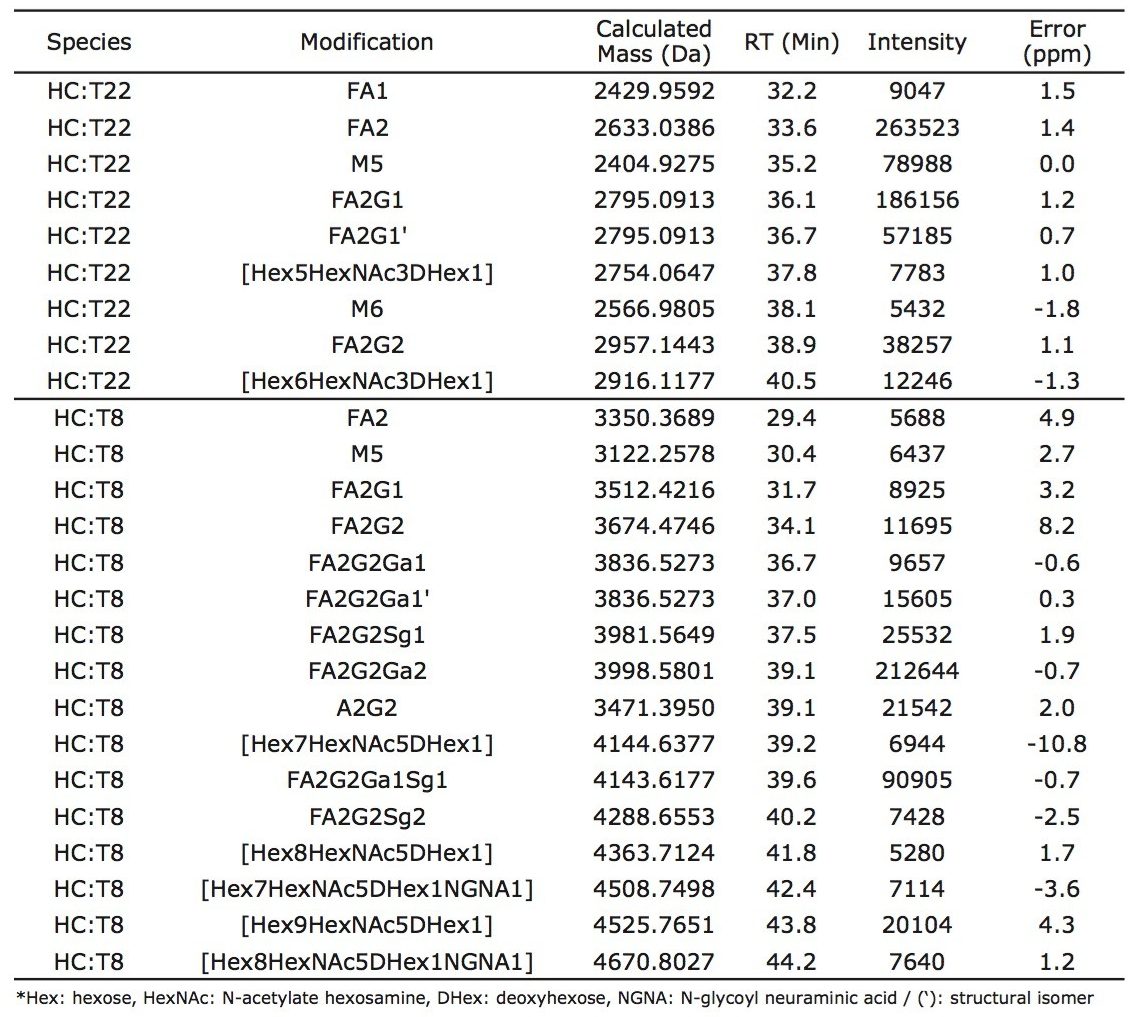
Lys-C digest of trastuzumab and NIST candidate reference material
An adaptation of a previously published single reaction vial, overnight (16+ hours) procedure4 was employed to prepare non-reduced Lys-C digests of trastuzumab and a IgG1K monoclonal antibody candidate reference material obtained from NIST (#8670, lot# 3F1b). TFA quenched digests were stored at -80 °C until analyzed. In preparation for HILIC chromatography, aqueous digests were diluted with 4 parts acetonitrile and 0.1 parts dimethylsulfoxide and were then centrifuged at 16 x 1000 g for 10 minutes to remove any insoluble composition. Supernatant from the centrifuged digest was thereafter injected.
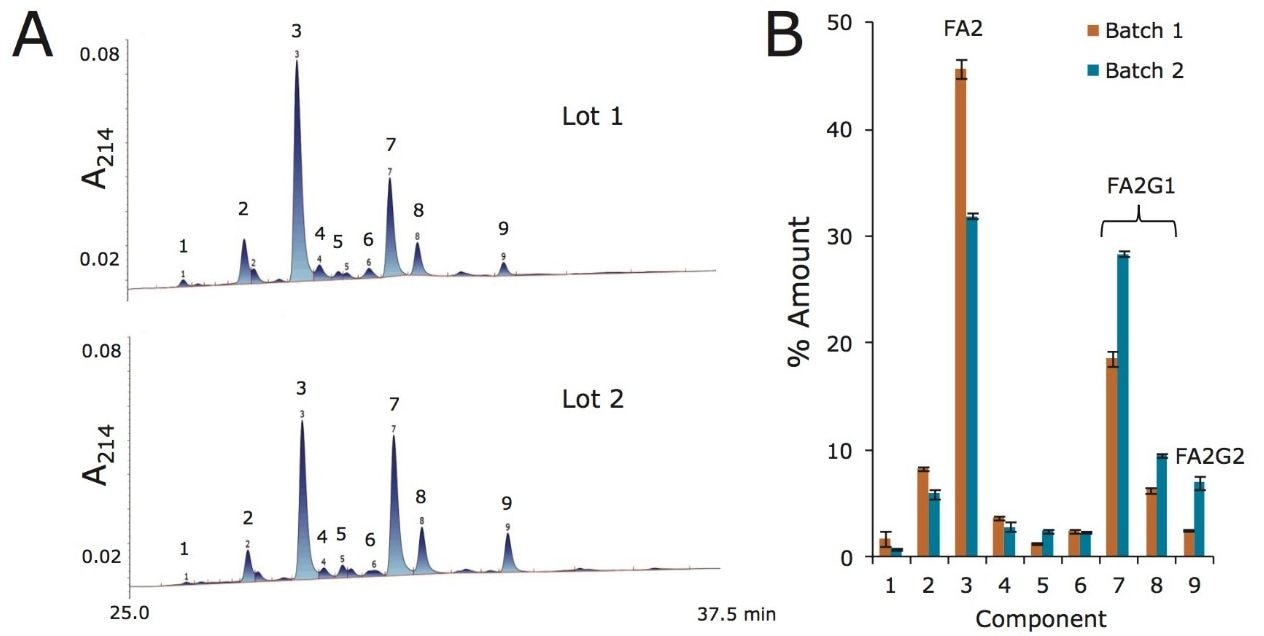
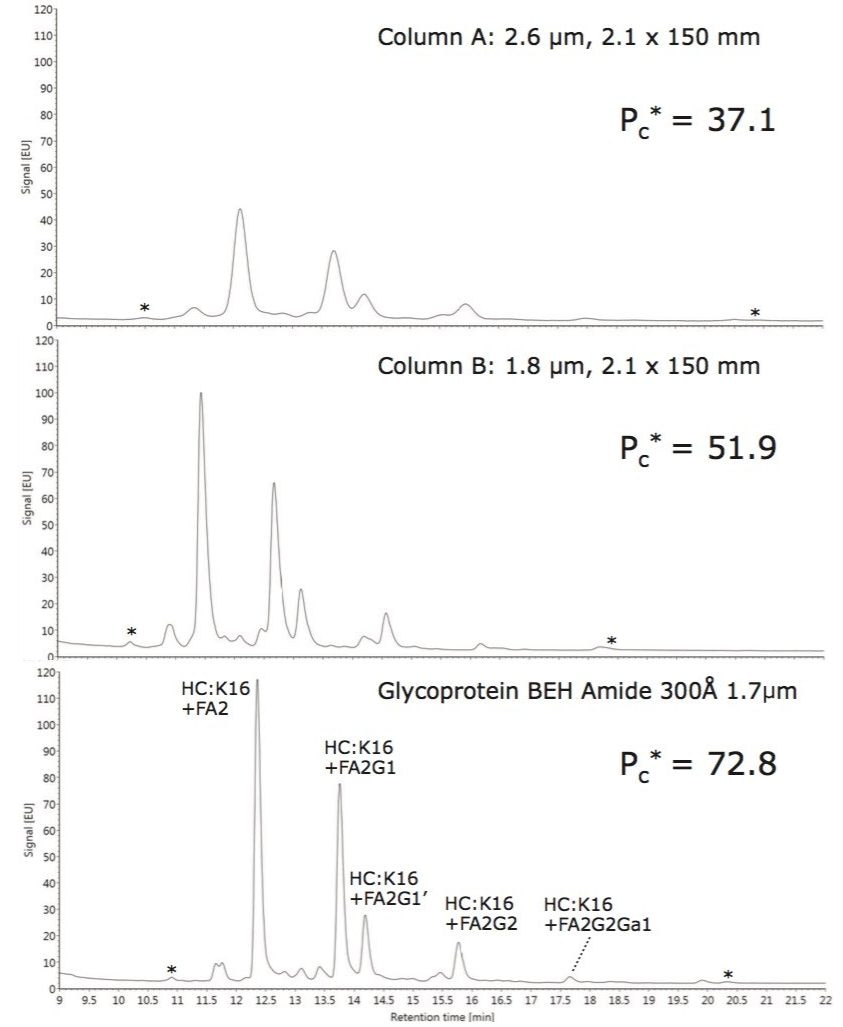
Lys-C/tryptic digest of cetuximab
Reduced and alkylated cetuximab was digested with a combination of Achromobacter protease I (Lys-C) and trypsin. Formulated cetuximab was concentrated to 10 mg/mL and buffer exchanged with a 10 kDa MWCO centrifugal filter (Millipore, Billerica, MA) into a solution of 6 M GuHCl, 50 mM DTT, and 0.2 M phosphate (pH 8.1), then incubated at 37 °C for 2 hours. Thereafter, the sample was diluted with a solution of iodoacetamide, bringing the antibody concentration to 8 mg/mL and the buffer composition to 4.8 M GuHCl, 40 mM DTT, 50 mM iodoacetamide, and 0.17 M phosphate (pH 8.1). Alkylation with iodoacetamide was allowed to proceed under these conditions for 10 min in the dark at 37 °C, before being quenched by the addition of cysteine, diluted with a urea- containing buffer, and mixed with Achromobacter protease I (Lys-C) at a 4:1 w/w ratio. The resulting digest solution of 0.8 mg/mL cetuximab, 0.5 M GuHCl, 3 M Urea, 40 mM NH2OH, 4 mM DTT, 5 mM iodoacetamide, 6 mM cysteine, and 0.1 M phosphate (pH ~7.1) was incubated at 37 °C. After 2 hours of incubation, this digest solution was diluted two fold with water and an aliquot of trypsin (Sigma T6567), such that the protein:trypsin ratio was 4:1 (w/w). After incubation at 37 °C for another 2 hours, the digest solution was again diluted two fold with water and a fresh aliquot of trypsin. With a total protein:trypsin ratio of 2:1 (w/w), the digest was left to incubate at 37 °C for 16 hours. Following this incubation, the digest was quenched by acidification with TFA and stored at -80 °C until analyzed. In preparation for HILIC chromatography, aqueous digests were diluted with 4 parts acetonitrile and 0.1 parts dimethylsulfoxide and were then centrifuged at 16 x 1000 g for 10 minutes to remove any insoluble composition. Supernatant from the centrifuged digest was thereafter injected.