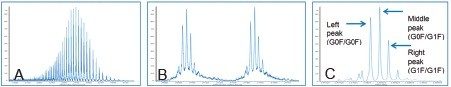
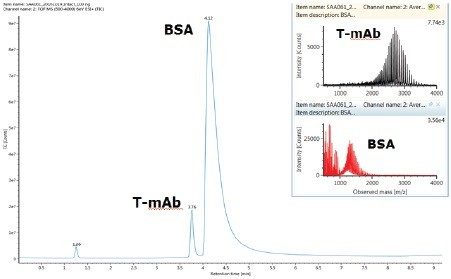
Biotherapeutics have emerged as one of the major sources of innovative medicines in drug discovery and development. As these biotherapeutic agents have complex structure and extremely high molecular weight, their detection and quantification can be challenging especially for the bioanalysis community that traditionally focuses on small molecules. The most common approach for quantifying these biotherapeutics is ligand-binding based assays complimented by LC-MS/MS analysis of surrogate peptides post protein digestion and sample cleanup. More recently, direct intact level protein quantification using LC-MS has gained traction as it offers direct measurement of proteins, including isoforms, and simpler sample preparation.
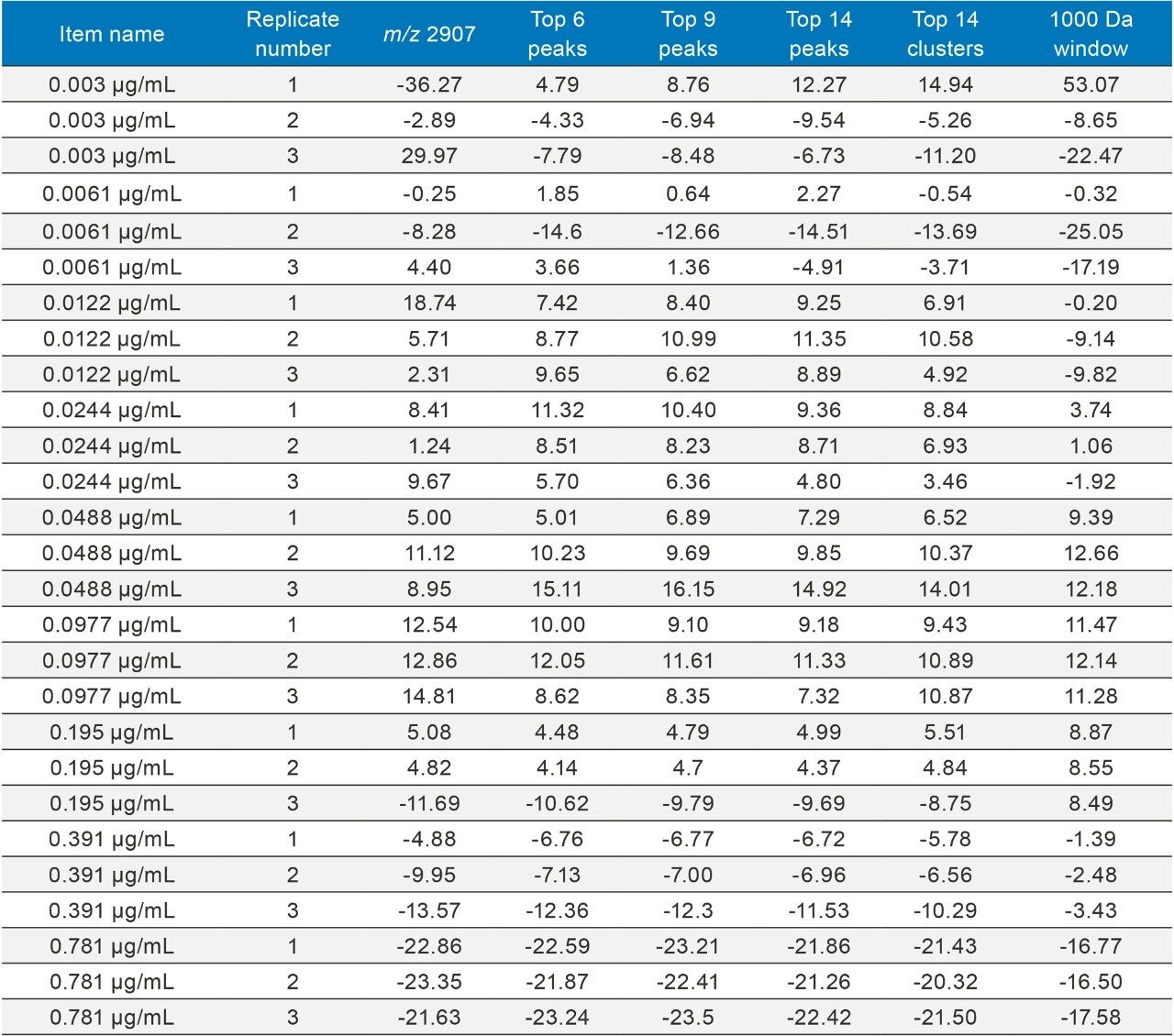
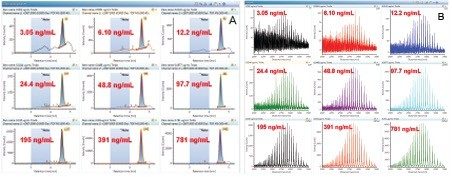
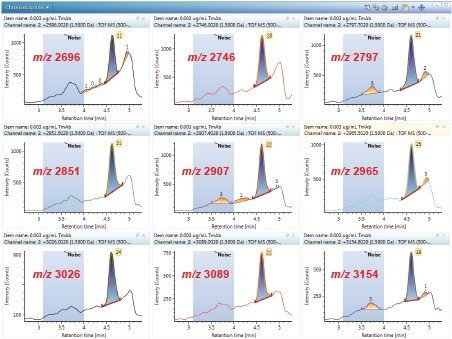
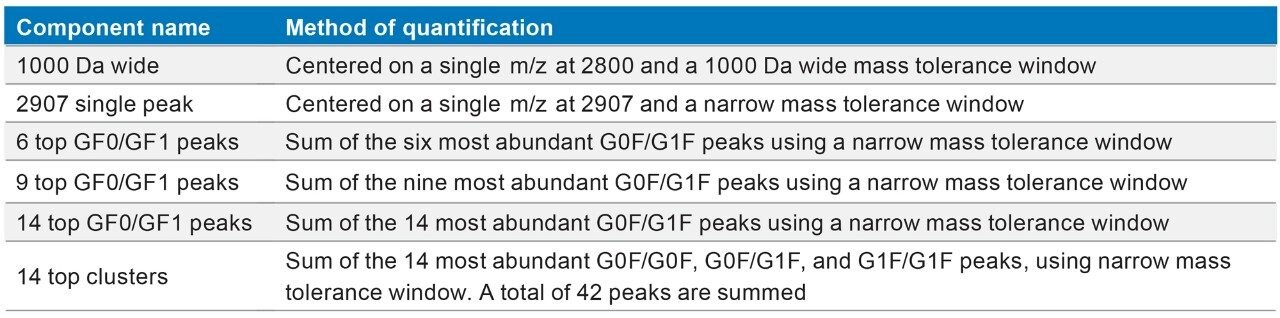
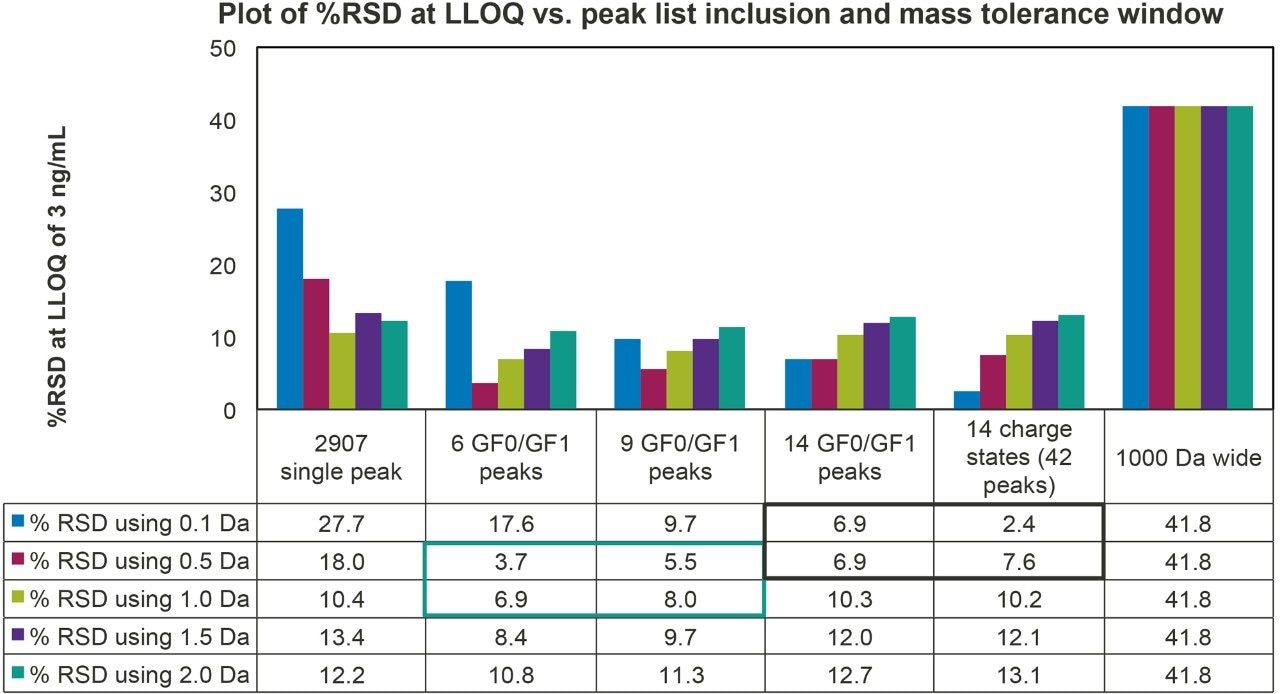
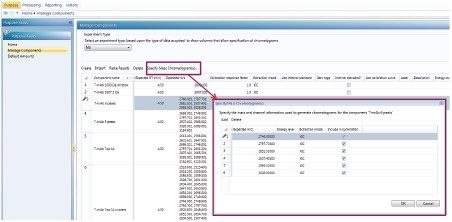
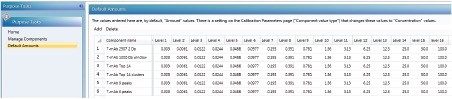
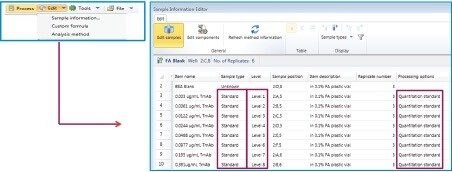
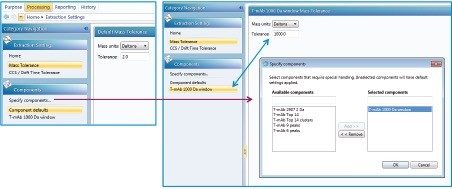
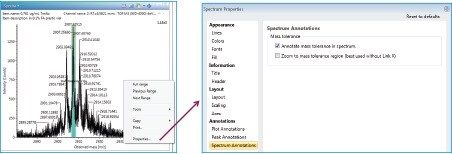
This application note describes a method for highly sensitive intact level quantification of a 150 kDa therapeutic protein, trastuzumab, in BSA solution capable of achieving an LLOQ of 3 ng/mL (30 pg load on column). The development and evaluation of peak based quantification is described in detail, from acquistion through processing. It provides detailed instruction for data processing options using UNIFI (using the Quantify Assay 2D Tof analysis method), including the impact on quantification of using different mass tolerance windows and numbers of peaks for integration of signal. Quantification attributes including: linearity, dynamic range, accuracy, limits of detection, and quantification are reported.