A Rapid, Workflow Driven Approach to Discovery Lipidomics Using Ion Mobility DIA UPLC/MS and Lipostar™
For research use only. Not for use in diagnostic procedures.
Abstract
We describe the use of an ion mobility-enabled high-resolution mass spectrometer combined with powerful, workflow driven informatics to characterize changes in the liver lipidome of the mouse following the administration of gefitinib, which is a selective inhibitor of epidermal growth factor receptor’s (EGFR) tyrosine kinase domain. The resulting data showed dysregulation of the lipid metabolism pathways with a time-related trajectory following the administration of the drug, with increased abundance of several LPCs and reduced presence of PCs in the dosed group. When interrogated versus a known cancer metabolic pathway the data suggested changes in the phosphocholine metabolic pathway.
Benefits
- Simple and robust acquisition strategy which:
- Generates high quality, information rich, and comprehensive data
- Allows for fast analysis
- Simple customizable workflow
- A flexible lipid identification approach that includes:
- A spectral matching approach
- High-throughput, bottom-up approach, and based on class-specific fragment ion recognition
- High-throughput identification of oxidized species
- Exploitation of m/z, CCS, and retention time values
- Various multivariate statistical analysis tools
- Quantification and pathway analysis tools
Introduction
Lipidomics is a rapidly growing area of omics science allowing researchers to probe changes in the lipidome as a result of disease, treatment, environmental exposure, lifestyle etc. The lipidome is complex, containing 1000’s of lipids from the polar free fatty acids, moderately polar bioactive lipids such as LPC, LPE, sphingomyelins, ceramides etc. to the nonpolar glycerol lipids such as triglycerides and cholesterol esters. Analysis of these lipids is challenging and, in a discovery mode, normally performed by a combination of reversed-phase liquid chromatography (RPLC) and accurate mass spectrometry. Despite developments in analytical technology the detection and identification of lipids of interest remains a significant challenge, mainly associated with data processing, database searching and lipid identification.
Here we show the key benefits of using UltraPerformance LC™ combined with ion mobility mass spectrometry and the powerful workflow driven Lipostar 2 software for the rapid, objective, and reliable discovery of compounds of interest. The benefits of this approach are demonstrated using the lipidomics data derived from the analysis of mouse liver tissue following the intravenous (IV) administration of Gefitinib, an epidermal growth factor receptor's (EGFR) tyrosine kinase domain used to treat various cancers. Here we highlight the key features and benefits of UPLC-IM-MS combined with Lipostar. The resulting data showed dysregulation of the lipid metabolism pathways with a time-related trajectory following the administration of the drug.
Experimental
Study Design
Mouse liver samples were sourced from a previous pharmacokinetics study of Gefitinib in male C57Bl/6JRj mice.1 The study was performed with full management review and according to national and EU guidelines by Evotec SAS (Toulouse, France). Briefly, Gefitinib was formulated in 0.5% Hydroxypropyl methylcellulose HMPC in 0.1% polysorbate 80 and administered intravenously (IV) at 10 mg/Kg. Liver samples were obtained pre-dose and at 0.5, 1, 3, 8, and 24 hour post-dose.
Sample Preparation
Lipids were extracted from the liver samples using the procedure outlined by Want et. al.2 Between 50–60 mg of tissue was sampled to 1.5 mL tubes pre-filled with silica beads. To this, 1 mL of dichloromethane/methanol (3:1, v/v) solution containing a 250-fold dilution of neat deuterated ceramide LIPIDOMIX™ (Avanti, Birmingham, Al, USA), SPLASH LIPIDOMIX™ (Avanti, Birmingham, Al, USA) and Gefitinib (d6) (Cayman Chemical, Ann Arbor, Michigan, USA) at 3000 ng/mL were added as internal standards. The tissue was homogenised before transferring the resulting organic layer to glass vials for drying under nitrogen. The extraction procedure was repeated on the remaining pellet to optimise the extraction.
After the extracts had dried down, samples were reconstituted in 1 mL of IPA/Acetonitrile (1:2, v/v) with ten minute sonication followed by a five minute centrifugation step to remove debris. The supernatant was transferred to maximum recovery vials for LC/MS analysis.
LC Conditions
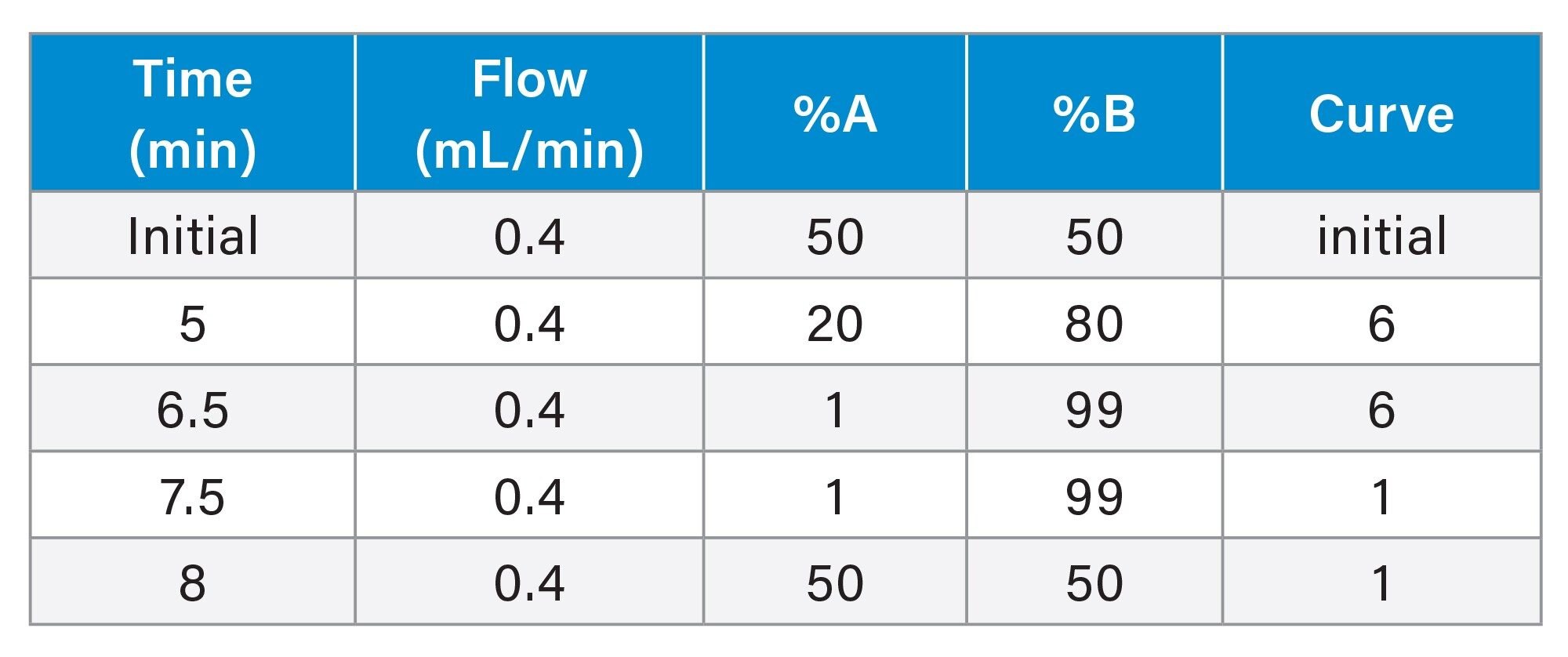
|
LC system: |
ACQUITY™ UPLC I-Class Flow Through Needle (FTN) |
|
Vials: |
Certified Glass Screw Neck Max Recovery Vials (p/n: 186000326c) |
|
Column(s): |
ACQUITY Premier UPLC CSH C18 2.1 x 100 mm, 1.7 µm (p/n: 186009488) |
|
Column temperature: |
55 °C |
|
Sample temperature: |
10 °C |
|
Injection volume: |
0.5 µL ESI+ and 2 µL ESI- |
|
Flow rate: |
0.4 mL/min |
|
Mobile phase A: |
60% ACN, 40% 10 mM ammonium acetate (v/v) |
|
Mobile phase B: |
90% IPA, 10% ACN |
Gradient Table
MS Conditions
|
MS system: |
Waters™ SYNAPT™ XS |
|
|
Ionization mode: |
ESI (+/-) |
|
|
Capillary voltage: |
2.5 kV (+); 1.7 kV (-) |
|
|
Acquisition mode: |
HDMSE |
|
|
Acquisition range: |
50–1200 Da |
|
|
Collision energy: |
Linear ramp (transfer CE) 25–45 eV |
|
|
Cone voltage: |
30 V |
|
|
Source temperature: |
150 °C |
|
|
Desolvation temperature: |
550 °C |
|
|
Cone gas flow: |
150 L/hr |
|
|
Desolvation flow: |
1000 L/hr |
Key TWIMS-MS Settings for Lipid Analysis
|
Helium cell gas flow: |
180.00 mL/min |
|
IMS gas flow (nitrogen): |
90.00 mL/min |
|
IMS wave velocity: |
650 m/s |
|
IMS wave height: |
40.0 V |
Results and Discussion
The process of lipid identification requires the development of a study design, sample preparation, analytical methodology, data acquisition, data componentization, post analysis statistics, database searching, drug metabolite identification (if appropriate), component review, biological aspect of the derived results, and finally reporting. Many approaches have been adopted to address this issue to improve throughput and accuracy. To this end HDMSE is an ideal solution as it provides, precursor ion, product ion, and collision cross section (CCS) information in single methodology. One of the biggest challenges in metabolomics and lipidomics is the accurate identification of “putative” biomarkers or indeed any of the components. Many initiatives have been developed to ensure the quality of these results such as the metabolomics quality assurance and quality control consortium (mQACC) or recent Lipidomic Standards Initiative (LSI).3,4,5 To streamline the process of data acquisition and analysis we have developed a workflow which combines the attributes of the ion mobility mass spectrometer, such as the SYNAPT XS or SELECT SERIES™ Cyclic™ IMS mass spectrometer, with data analysis powered by Molecular Discovery’s Lipostar software. The workflow is described below in Figure 1.
 Figure 1. Overview of the Lipostar workflow.
Figure 1. Overview of the Lipostar workflow.
Lipostar software is a comprehensive software tool for processing LC-MS/MS-based lipidomic data, which can incorporate multiple MS acquisition types such as data dependent (DDA) and data independent (DIA) acquisition datasets, including both ion mobility (HDMSE) and SONAR. The Lipostar software solution facilitates raw data import, peak detection, lipid identification, quantification, statistical analysis, trend analysis, flux analysis, and biopathways analysis. It is applicable to both untargeted and semi-targeted experiments, including stable isotope labelling studies. Within a Lipostar analysis session, different modes of lipidomics analysis can be combined to increase knowledge and obtain a more comprehensive analysis of lipid profiles. To illustrate the power of the combination of the Waters UPLC-IM-MS System combined with the Lipostar application software this platform was used for the analysis of liver tissue extract following the intravenous (IV) administration of gefitinib to male C57Bl/6JRj mice. Typical composite positive and negative ion lipid chromatograms are shown in Figure 2.
Peak Picking/Feature Finding
Waters data files (.raw) generated using high-resolution instruments were directly imported into the most recent version of Lipostar, Lipostar 2. The “LC-MS Settings” window allows the user to either select the default setting according to the LC-MS instrument used or customize and save a new setting.
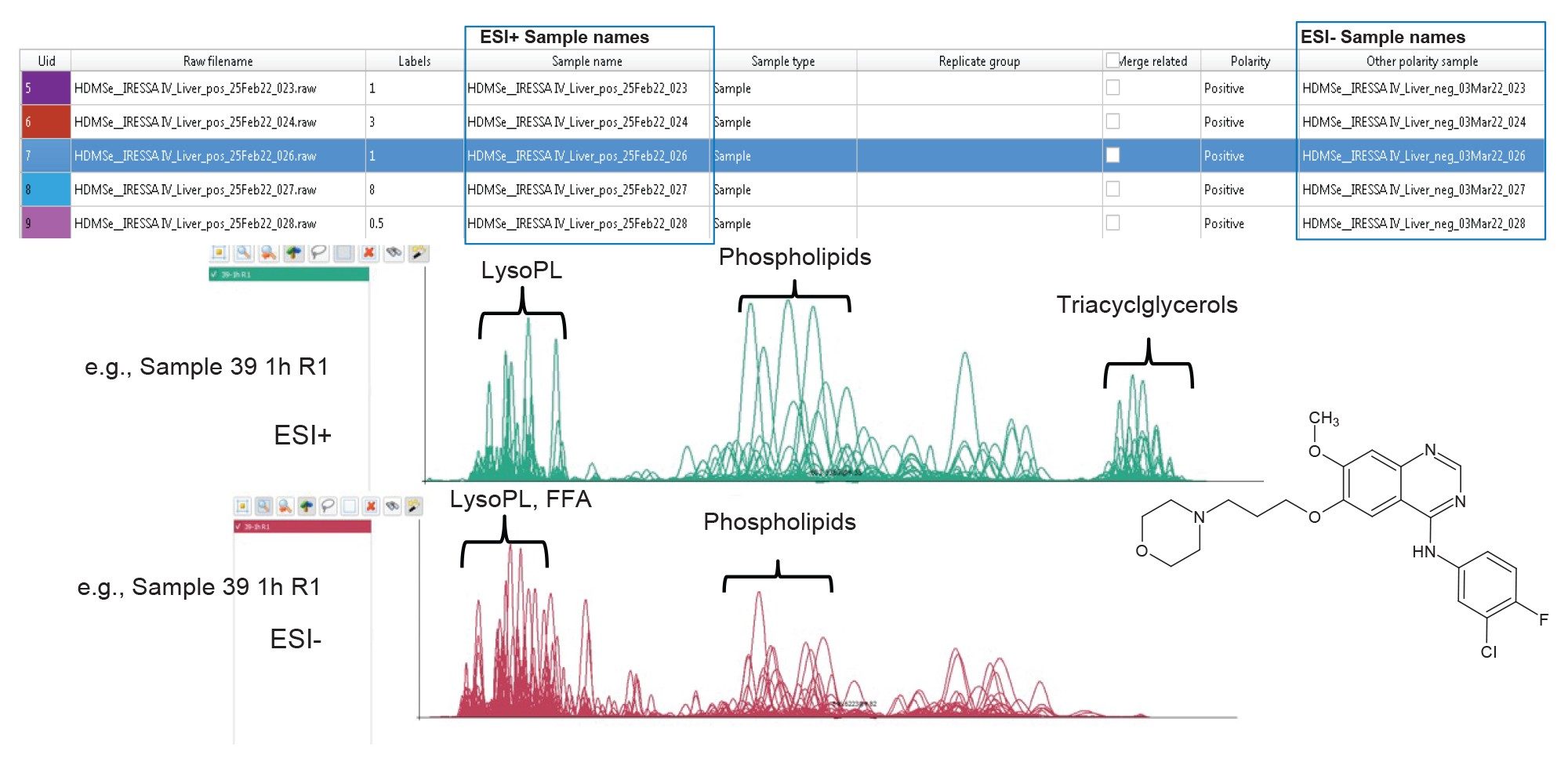 Figure 2. Data from positive mode and negative mode analysis of the Gefitinib mouse liver extracts can be imported to the same session.
Figure 2. Data from positive mode and negative mode analysis of the Gefitinib mouse liver extracts can be imported to the same session.
The informatics solution allows data files from positive and negative polarity modes to be added to the same analysis session, with labels assigned manually or using correctly formatted .csv files for sample comparison and multivariate statistics. Figure 2 represents an exemplar dataset, showing the importation of positive and negative ion data relating to the liver lipid extracts from the gefitinib study. The chromatographic conditions used for both polarity modes are identical, therefore allowing for convenient, easy comparison and ultimately providing improved confidence in the identifications provided. Lipostar performs data processing in five steps: (1) baseline and noise reduction, (2) peak extraction; (3) smoothing; (4) signal-to-noise ratio; and (5) deisotoping and deconvolution[6].
Alignment and Statistics
Untargeted lipidomics typically involves comparing lipid profiles from several samples from a large batch. Following peak extraction, data processing involves the lipid profile of each analytical sample being initially defined as a series of mass-to-charge ratios at given retention times (m/z@tR); at this stage, isotopic peaks of a lipid as well as peaks related to different adducts are not clustered and hence treated as distinct entities. The defined lipid profiles are then aligned to form a data matrix, where rows represent the analyzed samples and columns represent the m/z@tR entries as shown in Figure 3. Following alignment, a column-wise search for isotopic patterns is performed for peaks which possess the same tR, these peaks are then simplified to one entry.
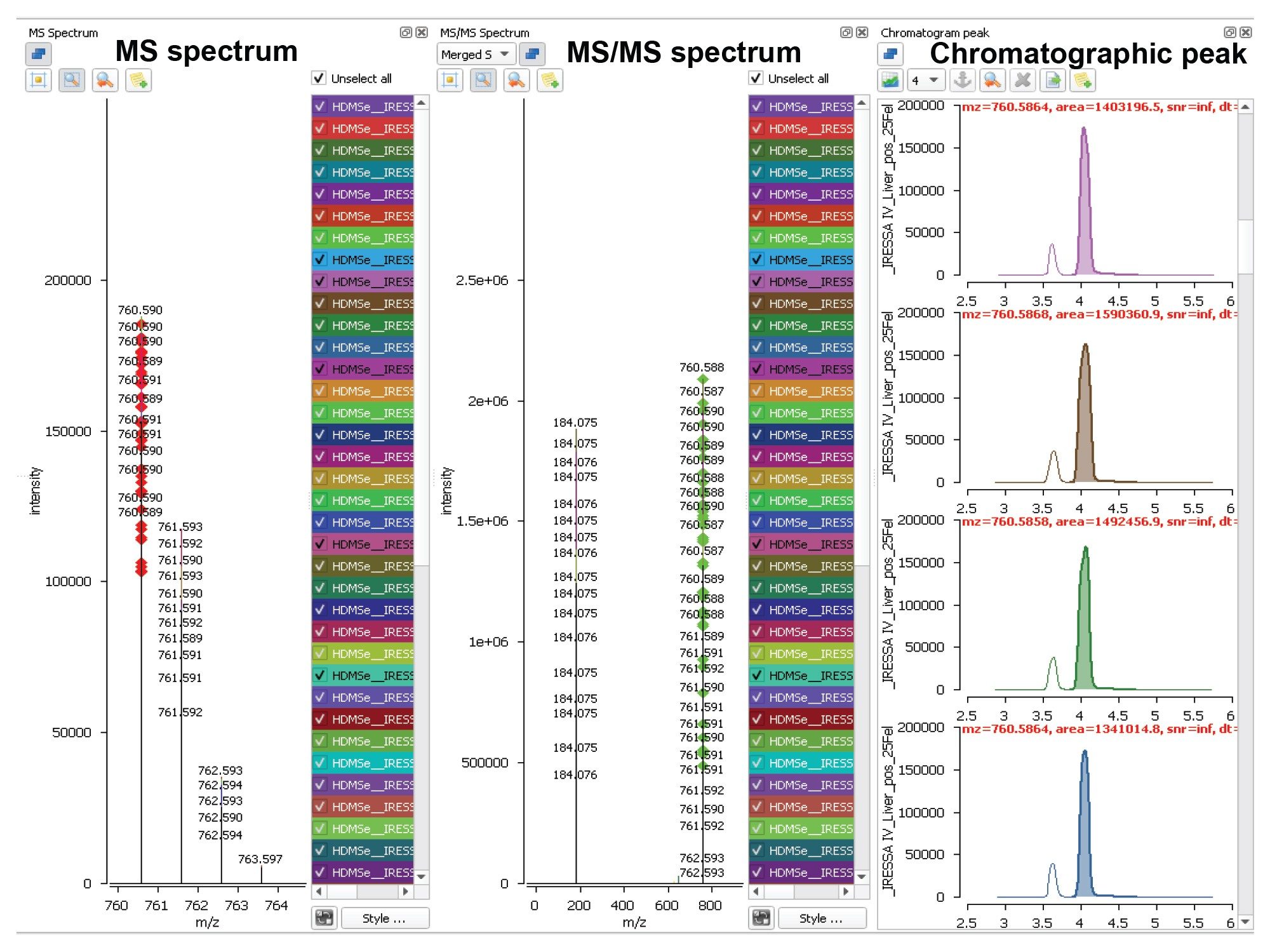 Figure 3. An example from the mouse liver data matrix relating to the feature 760.5879@4.14 (m/z@tR), which shows good alignment of the merged MS and MS/MS spectra across the samples of the analysis.
Figure 3. An example from the mouse liver data matrix relating to the feature 760.5879@4.14 (m/z@tR), which shows good alignment of the merged MS and MS/MS spectra across the samples of the analysis.
The resulting mass/charge – retention time pair feature table can then be subject to multivariate analysis (MVA) to reveal any underlying patterns in the data. Various unsupervised (e.g., principal component analysis: PCA, Consensus PCA) and supervised (e.g., partial least-squares: PLS, PLS-DA, O-PLS) algorithms for multivariate analysis are available within the Lipostar software. In Figure 4 we show the resulting MVA data obtained from the PLS-DA model and S plot using the negative mode data. This data shows a clear time-related trajectory of the data using a combination of endogenous lipids and drug-related metabolites. Further advanced tools for the visualization and interpretation of the statistical analysis, as well as the evaluation of robustness, are also possible. For instance, prior to selecting variables (i.e., lipids) to be used for a statistical purpose, the frequency plot (frequency of variable occurrence vs the number of samples) can be employed.
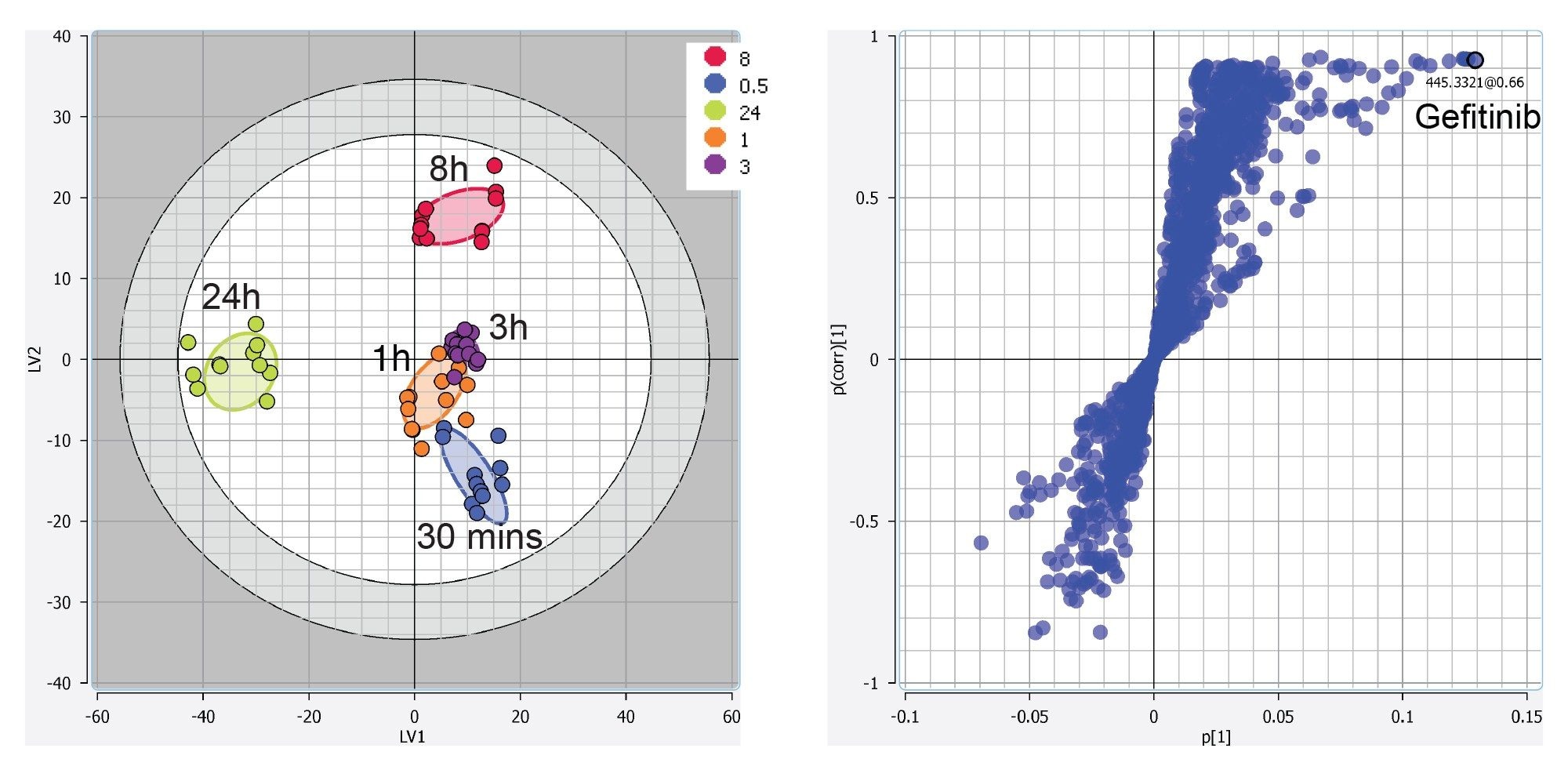 Figure 4. Example statistical tools available within Lipostar. Here we show the negative mode PLS-DA LV1 vs. LV2 score plot (left) and S plot (right) from the liver extract samples at various time points after dosing with the Gefitinib drug.
Figure 4. Example statistical tools available within Lipostar. Here we show the negative mode PLS-DA LV1 vs. LV2 score plot (left) and S plot (right) from the liver extract samples at various time points after dosing with the Gefitinib drug.
Identification and Quantification
As previously stated, the accurate identification of putative lipids is extremely challenging, with databases being used extensively. A description of the Lipostar identification workflow is discussed in detail by Goracci et al.3 Briefly within Lipostar, lipids are generally identified by importing or building a database of lipid structures through the DB Manager module. In addition to the exact mass of the lipid, the user can import additional information, such as retention time and CCS values. For Waters files processed in Lipostar, the Waters Lipidomic Profiling CCS library can be imported, when available. Finally, lipid structures in the database can be fragmented by a rule-based approach in the DB Manager module. Based on experimental evidence from literature or in house data, several fragmentation rules are labelled according to their importance in lipid annotation. For example, a fragmentation rule can be labelled as “mandatory” (e.g., the rule that define the fragment with m/z 184.074 for the protonated diacylglycerophosphocholines), or “recommended” (referring to a fragment match with a significant contribution to the final score, but not per se enough to unequivocally determine the lipid annotation). The generated fragments are also associated to weights (tunable by the user based on the instrumental conditions used).
A scoring function that is a combination of the difference between experimental and theoretical mass (Δ ppm), MS/MS and CCS values from databases (when available) is associated to each identification result. The weight to be assigned to each partial score can be set by the user when the identification method is defined (e.g., for full scan acquisition, the MS/MS score can be set at zero). In addition, based on the level of confidence for an identification result, especially based on MS/MS fragmentation matches, identified features are colour coded to provide an indication of the reliability and robustness of the identifications (Figure 5), which comprise of:
- Dark Green: the first proposed match is of high-confidence and based on a good MS/MS spectral matching.
- Orange: the first proposed match is an identification awaiting approval/review since the sum composition or lipid class conflicts or MS/MS information is not available.
- Red: not identified.
Concerning the dark green labelled results, “mandatory” and “recommended” fragmentation rules must be satisfied. Although identification within Lipostar is mainly run by using a lipid database, a database-free identification algorithm can also be applied for class or subclass annotation level, when MS/MS data are available; this approach is based on collections of class specific fragments embedded in the software.
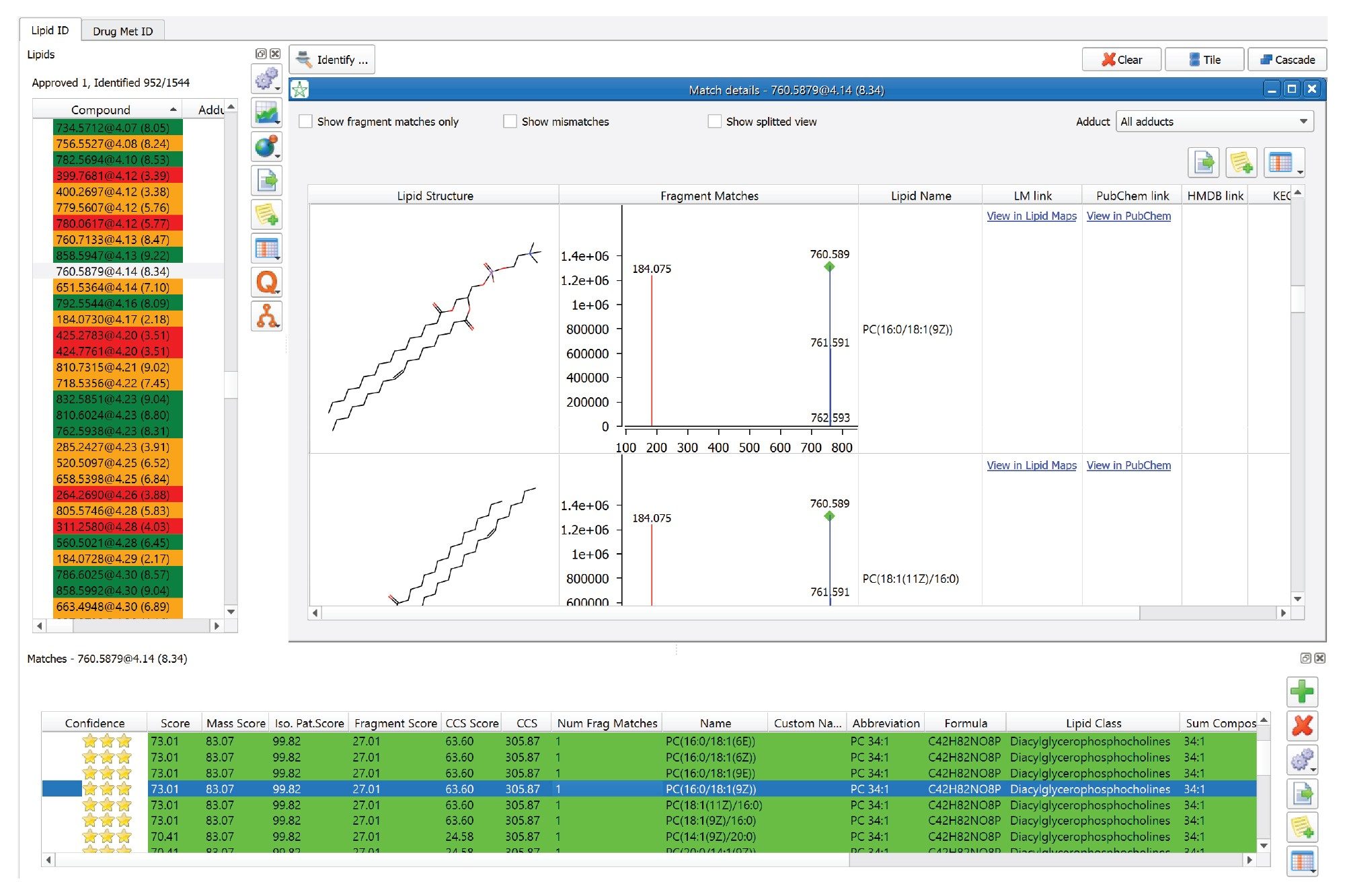 Figure 5. Shows an example of a high-confidence identification. In this example feature, 760.5879@4.14 is identified as PC (34:1). The MS/MS spectrum is available and all the components (i.e., acyl chains) of the proposed lipid are readily identified (dark-green label).
Figure 5. Shows an example of a high-confidence identification. In this example feature, 760.5879@4.14 is identified as PC (34:1). The MS/MS spectrum is available and all the components (i.e., acyl chains) of the proposed lipid are readily identified (dark-green label).
When the identification approach based on a lipid database is used, unidentified m/z entries are further processed to identify potential oxidized forms, based on the exact mass comparison, and on a preliminary inspection of the MS/MS spectra.6
Lipostar also contains a simplified version of the Mass-MetaSite algorithm to automatically detect drugs and their metabolites.7 The search of these species in the samples set is very useful when the variation in the lipidome upon drug treatment is monitored, especially in the case of lipophilid drugs, which could be extracted together with lipids. Starting from the chemical structure of a drug as input file, the structures of potential metabolites are generated by applying in silico common metabolic reactions. For each predicted metabolite, a fragment library is automatically generated and metabolites in the samples set are identified by a spectral matching approach.
In this study, the Mass-MetaSite algorithm in Lipostar was used to identify the drug-related metabolites of gefitinib formed in vivo, and these species were removed from the analysis to establish changes associated with the endogenous lipid levels only. Figure 6 shows an example of a gefitinib metabolite identified among the detected features by applying the Mass-MetaSite algorithm in Lipostar. This metabolite having m/z value of 421.1441 has been previously reported by Molloy et al. as metabolite M6.1
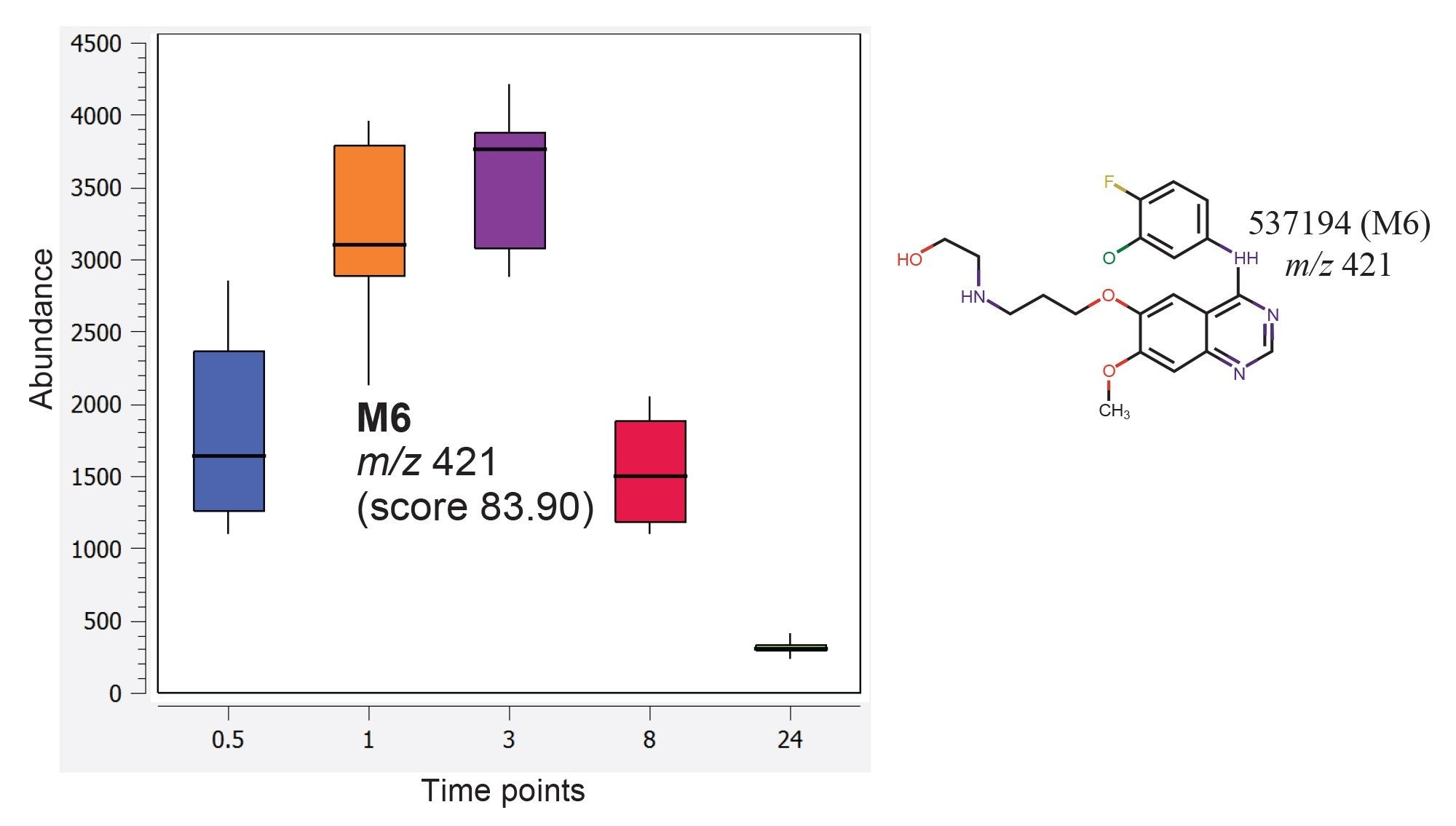 Figure 6. M6 metabolite (C20H22ClFN4O3) was predicted using Mass-MetaSite. Box and whisker plots of the relative abundance of this metabolite from the mouse liver extracts at various time points are shown.
Figure 6. M6 metabolite (C20H22ClFN4O3) was predicted using Mass-MetaSite. Box and whisker plots of the relative abundance of this metabolite from the mouse liver extracts at various time points are shown.
Following statistical analysis of the data sets and lipid identification, determination of lipid concentration is often the next task. The Lipostar quantification tool can also be used to quantify endogenous lipids in both targeted and untargeted experiments. Lipostar allows the lipid scientist either to select the concentrations corresponding to the linearity range to refine the linear regression, or to apply the polynomial regression to the entire interval. Calibration curves for the various adducts of the calibrant can also be generated.
The LOQ and the LOD is calculated using the formula:
LOQ = 10 * Standard Error / slope
LOD = 3.3 * Standard Error / slope
Where the standard error uses only the enabled points of the computed regression.
Data Interpretation
Trend analysis is a powerful tool allowing lipid abundances to be grouped by anticipated trends. In addition, the application of clustering algorithms and the “group by trend” features, enables the identification of potential biomarkers but also the inspection of new adducts and in-source fragments. Figure 7 shows how two closely related lipid species (i.e., LPCs and PCs) show distinct differences across the gefitinib time course study.
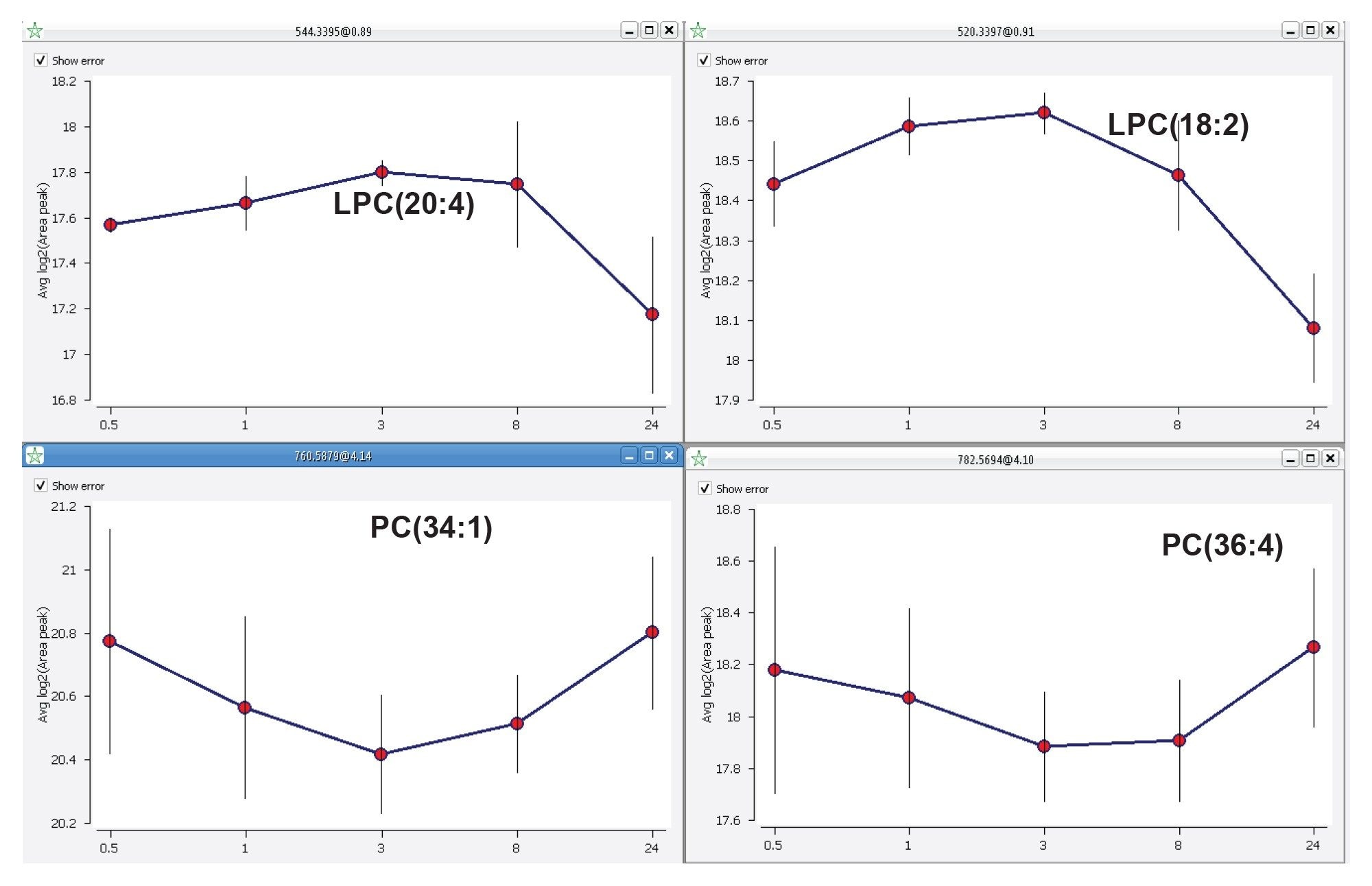 Figure 7. Lipid abundance trend examples: LPCs with similar abundance follow over various time points (top) and the trend key PC species follow over the same time points of liver extract samples after dosing with a drug (bottom).
Figure 7. Lipid abundance trend examples: LPCs with similar abundance follow over various time points (top) and the trend key PC species follow over the same time points of liver extract samples after dosing with a drug (bottom).
Pathways
The ability to map lipids to pathways is biologically significant, allowing for mechanistic and process interpretation. Lipostar has a collection of lipid pathway maps (i.e., global, metabolic and disease) that can be linked either to the database of fragmented lipids or to the results of a lipid identification. Figure 8 shows a portion of the metabolic pathway depicted in “Prostate Cancer” disease map. The cancer-related pathway was used since gefitinib was developed for treating lung cancer. For example, PC (34:1), which is a phosphocholine lipid and thus being projected in the PC node, is directly linked to the LPC1 node (monoacylglycerophosphocolines), which are known to decrease their concentration in Prostate cancer (highlighted in blue). Contrarily, elevated levels of other lipids (highlighted in red) are also shown to be indirectly linked.
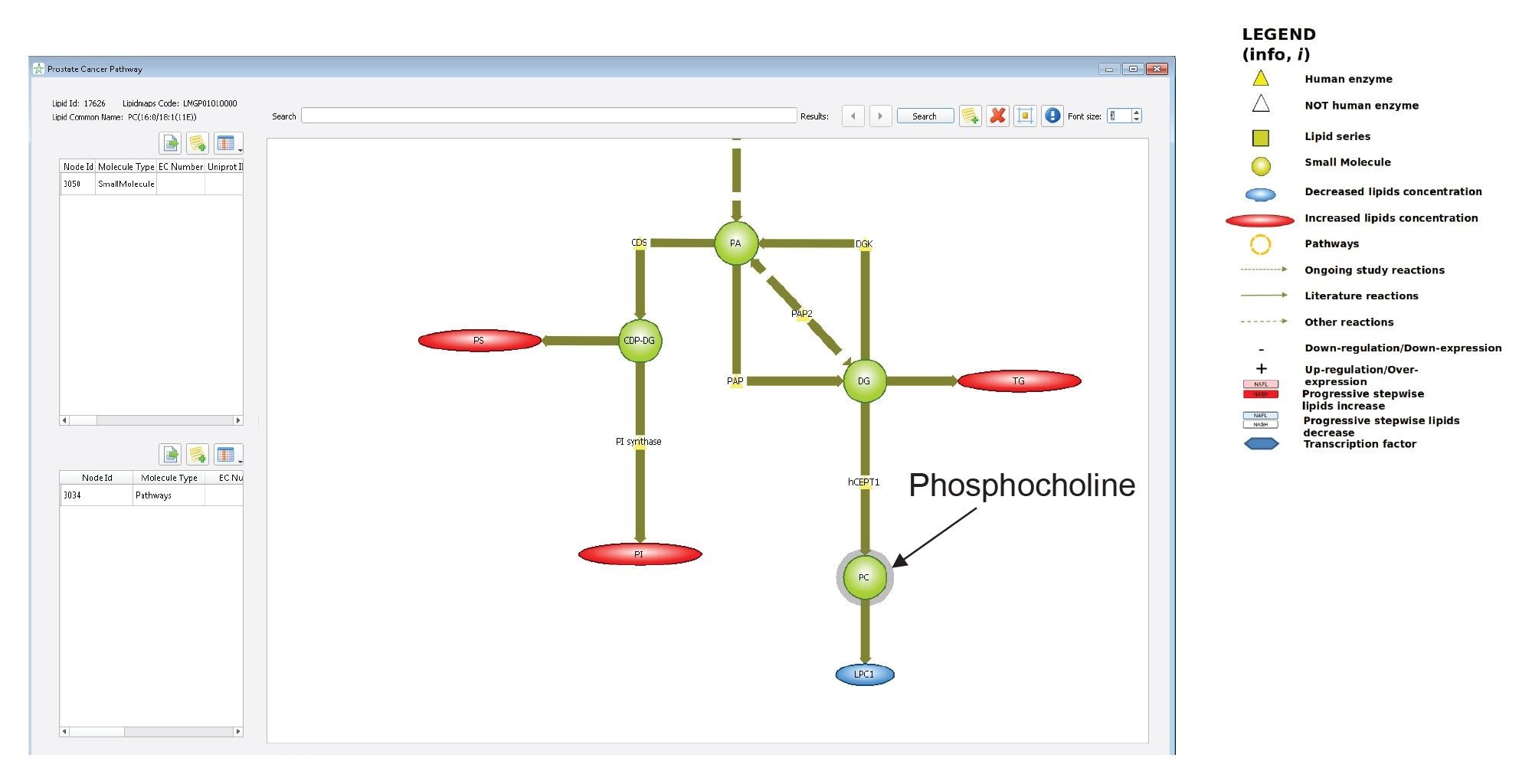 Figure 8. Portion of the metabolic pathway depicted in “Prostate Cancer” disease map in Lipostar. The node that includes PC(34:1), which was shown to be a significant contributor to the multivariate models of the gefitinib mouse liver extracts, is highlighted. Associated lipids in red were found to increase in concentration in protate cancer according to literature, whilst those in blue are expected to decrease.
Figure 8. Portion of the metabolic pathway depicted in “Prostate Cancer” disease map in Lipostar. The node that includes PC(34:1), which was shown to be a significant contributor to the multivariate models of the gefitinib mouse liver extracts, is highlighted. Associated lipids in red were found to increase in concentration in protate cancer according to literature, whilst those in blue are expected to decrease.
The pathways maps can be also linked to the identification results. Lipostar also allows displaying of all the approved compounds in the maps simultaneously.
Conclusion
The combination of reversed–phase UPLC, ion mobility enabled high-resolution mass spectrometry and an intelligent workflow driven software allows for the rapid accurate lipidomic analysis of biological samples. The Lipostar software enables accurate quantification and identification of compounds using database searches, Waters LC-MS datasets (DDA or DIA-based), including ion mobility enabled MS can be directly processed. The user-friendly, easy-to-use workflow described above allowed for the rapid processing of lipidomic data such as that generated for the gefitinib study described here. The data generated from the gefitinib study was subjected to extensive statistical analysis using the tools available within the Lipostar 2 software to enable biological interpretations. The pathway tools within the software were used to pinpoint the metabolic pathways most affected by lipid perturbation.
Acknowledgements
We would like to thank Paolo Tiberi (Molecular Discovery Ltd, Hertfordshire UK) and Laura Goracci (University of Perugia, Perugia, Italy) for their time and effort.
References
- Molloy B.J, King A, Mullin L.G, Gethings L.A, Riley R, Plumb R.S, Wilson I.D. Rapid Determination of the Pharmacokinetics and Metabolic Fate of Gefitinib in the Mouse Using a Combination of UPLC/MS/MS, UPLC/QToF/MS, and Ion Mobility (IM)-enabled UPLC/QToF/MS. Xenobiotica 2021, 51, 434–446.
- Want, E., Masson, P., Michopoulos, F. et al. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat Protoc, 2013, 8, 17–32. https://doi.org/10.1038/nprot.2012.135.
- Beger RD, Dunn WB, Bandukwala A, Bethan B, Broadhurst D, Clish CB, Dasari S, Derr L, Evans A, Fischer S, Flynn T, Hartung T, Herrington D, Higashi R, Hsu P-C, Jones C, Kachman M, Karuso H, Kruppa G, Lippa K, Maruvada P, Mosley J, Ntai I, O'Donovan C, Playdon M, Raftery D, Shaughnessy D, Souza A, Spaeder T, Salholz B, Tayyari F, Ubhi B, Verma M, Walk T, Wilson I, Witkin K, Bearden DW, Zanetti KA. Towards quality assurance and quality control in untargeted metabolomics studies. Metabolomics. 2019 Jan 3; 15(1): 4.
- Evans AM, O’Donovan C, Playdon M, Beecher C, Beger RD, Bowden JA, Broadhurst D, Clish CB, Dasari S, Dunn WB, Griffin JL, Hartung T, Hsu P-C, Huan T, Jans J, Jones CM, Kachman M, Kleensang A, Lewis MR, Monge ME, Mosley JD, Taylor E, Tayyari F, Theodoridis G, Torta F, Ubhi BK, Vuckovic D on behalf of the Metabolomics Quality Assurance, Quality Control Consortium (mQACC). Dissemination and analysis of the quality assurance (QA) and quality control (QC) practices of LC-MS based untargeted metabolomics practitioners. Metabolomics. 2020 Oct 12; 16(10): 113.
- O’Donnell, V. B., Ekroos, K., Liebisch, G. & Wakelam, M. Lipidomics: Current state of the art in a fast-moving field. Wiley Interdiscip. Rev. Syst. Biol. Med. 12, 1–6 (2020).
- Laura Goracci, Sara Tortorella, Paolo Tiberi, Roberto Maria Pellegrino, Alessandra Di Veroli, Aurora Valeri, and Gabriele Cruciani (2017) LipoStar, a Comprehensive Platform-Neutral Cheminformatics Tool for Lipidomics, Analytical Chemistry 2017 89 (11), 6257–6264, DOI: 10.1021/acs.analchem.7b01259.
- Zamora I, Fontaine F, Serra B, Plasencia G. High-throughput, computer assisted, specific MetID. A Revolution for Drug Discovery. Drug Discov Today Technol. 2013 Spring;10(1): e199-205. doi: 10.1016/j.ddtec.2012.10.015. PMID: 24050248.
720007817, December 2022