Vergleich der Präzision von Gerät zu Gerät: Millimasseinheiten (mmu), Messfehler (ppm) und Auflösung
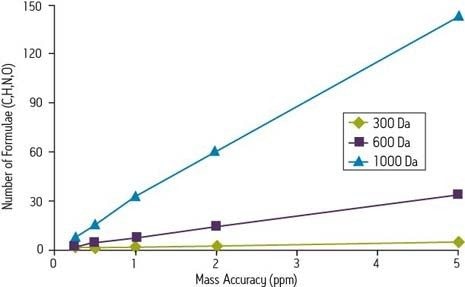
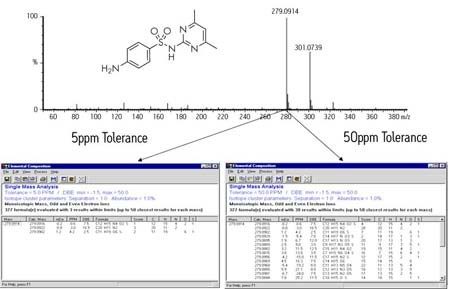
Laut dem Accurate Mass Best Practice Guide des VIMMS-Programms, einer Initiative des britischen National Measurement Systems, können die meisten für akkurate Massenmessungen verwendeten Geräte eine Genauigkeit von 10 ppm oder besser erreichen.
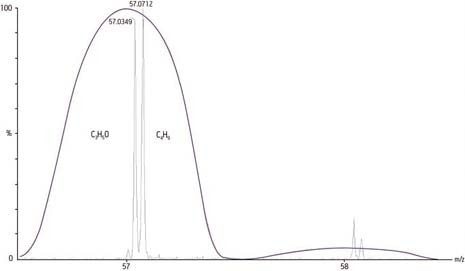
Eine berechnete Masse von 118 Da, die von einem modernen Massenspektrometer mit einer Genauigkeit von 2 mmu gemessen wird, würde einen Fehler von 17 ppm aufweisen, was nach heutigen Maßstäben für die eindeutige Bestimmung einer chemischen Formel dieser Masse ausreicht:
Monoisotopisch berechnete exakte Masse = 118 Da
Gemessene genaue Masse = 118,002 Da
Differenz = 0,002 mmu
Fehler [Differenz/exakte Masse x 106] = 17 ppm
Ein Gerät, das bei 750 m/z reagieren kann, ebenfalls mit einem Mangel von 2 mmu, wäre auf 2,7 ppm genau. Im ersten Fall ist die Messung mehr als ausreichend für die eindeutige Identifizierung einer chemischen Formel, entsprechend den veröffentlichten Standards des Journal of The American Society for Mass Spectrometry. Im letzteren Fall ist die Messung jedoch nicht präzise genug. Nur die Fourier-Transformations-Ionenzyklotronresonanz-Massenspektrometrie (FTICR) höchster Ordnung kann eine solche Präzision bei höheren Massen erreichen.
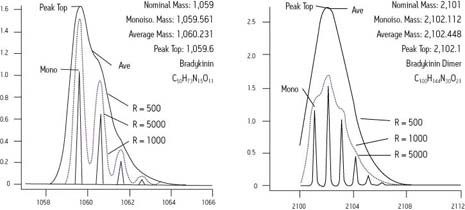
Eine umfassende Methode zur Bewertung der Genauigkeit der Massenmessung eines Geräts, die der beabsichtigten Verwendung entspricht, ist die Berechnung des quadratischen Mittelwerts oder RMS-Fehlers. Zur Veranschaulichung der Anwendung wird im Folgenden die Spezifikation der Massenmessgenauigkeit eines handelsüblichen ToF-Massenspektrometers verwendet.
„Die Massenmessgenauigkeit des Geräts ist unter normalen Betriebsbedingungen besser als ein bestimmter ppm RMS über den bestimmten m/z-Bereich, basierend auf einer Anzahl von aufeinanderfolgenden Wiederholungsmessungen eines Analytpeaks (mit bestimmtem m/z), unter Verwendung eines geeigneten Referenz-Peaks (mit bestimmtem m/z). Analyt- und Referenz-Peaks müssen eine ausreichende Intensität aufweisen und frei von Störungen durch andere Massen sein.“
Es gibt einige wichtige Punkte und Annahmen, die berücksichtigt werden müssen:
- Eine Gerätekalibrierung wurde bereits mit Peaks bekannter Masse unter Verwendung eines Kalibrierstandards durchgeführt. Der Referenz-Peak wird verwendet, um etwaige Schwankungen der Gerätekalibrierung im Laufe der Zeit auszugleichen, und die Genauigkeit der Massenmessung wird anhand des Peaks des Analyten bestimmt.
- Normale Betriebsbedingungen – können auch Angaben zu chromatographischen Bedingungen (für LC/MS-Leistungsdaten) und allen damit verbundenen MS-Betriebsbedingungen (z. B. Massenauflösung, interessierendes m/z oder spektrale Aufnahmerate) enthalten.
- Ausreichende Intensität – setzt voraus, dass die Ionenzahl die Charakterisierung der (Massenmessungs-)Genauigkeit und Präzision des betreffenden Geräts nicht beeinträchtigt. Zu wenige Ionen führen zu einer schlechten Ionenstatistik und zu viele Ionen können zu einer Sättigung des Detektors führen. Beides führt zu einer größeren Schwankung der Standardabweichung bei Wiederholungsmessungen und beeinflusst die Berechnung des RMS-Fehlers (auch für die Kalibrierung des Geräts relevant) negativ.
- Störungsfrei – geht davon aus, dass die Massenmessung des Peaks mit bekannter Masse frei von Störungen durch Ionen gleicher oder ähnlicher Masse ist. Überlappende Peaks führen zu einer schlechten Genauigkeit der Massenmessung, was sich auch nachteilig auf die korrekte Charakterisierung der Genauigkeit oder Präzision des Geräts auswirkt (auch relevant für die Gerätekalibrierung).
- Die Referenz ist eine gute Darstellung des m/z-Bereichs, der für die Analyse eines bestimmten Probentyps relevant ist.
Der RMS-Fehler wird anhand der folgenden Beziehung berechnet, wobei Eppm der ppm-Fehler und n die Anzahl der betrachteten Massen ist: