提高效率:使用ACQUITY™ QDa™ II质谱检测器快速可靠地分析制药辅料中的亚硝酸盐含量
摘要
优势
- 通过全面的29天分析,展示ACQUITY™ QDa™ II质谱检测器在常规测定典型制药辅料中亚硝酸盐含量方面的应用性能
- 重点介绍仪器的易用性,为操作人员提供有关维护间隔、记录任何仪器停机时间,以评估仪器在高样品通量实验室中的适用性
- 评估在常规分析检测中定量µg/g水平亚硝酸盐的方法精度,评估系统在应用工作流程中的可靠性
- 采用Empower™色谱数据系统(CDS)软件,确保结果报告的合规性和全面性
简介
近年来,药品中N-亚硝胺的存在迅速成为制药行业发展中备受关注的问题。全球监管机构已对多种药物类别中的N-亚硝胺污染提出了安全担忧,因为有观点认为,患者长期暴露于高于安全水平的此类杂质可能会面临致癌和致突变风险1,2。 因此,降低风险并控制这些化合物的存在已成为药品开发和质量评估中不可或缺的部分。
亚硝酸盐是N-亚硝胺形成的最关键风险因素3。 在酸性条件下,亚硝酸盐通常与仲胺或叔胺反应形成活性NO+载体物质,作为亚硝化剂。在特定条件下,这种亚硝化剂与不稳定的胺发生无差别相互作用,将显著影响最终药物产品中所有可能存在的N-亚硝胺的水平。因此,筛查总亚硝酸盐含量,作为此类形成的单一潜在前体,可以作为支持风险评估的简单且实用的方法,从而有效降低形成N-亚硝胺的风险。这样将有助于减少执行传统亚硝胺分析的需求,由于灵敏度要求,传统分析通常更具挑战性。
亚硝酸盐污染通常来自原料、中间体、辅料,以及合成过程中使用的溶剂和试剂。然而,辅料中作为杂质存在的亚硝酸盐浓度通常以百万分之一(ppm)计量,并且是亚硝化剂的主要来源4,5。 众所周知,辅料中的亚硝酸盐水平在不同类型和供应商之间存在着显著差异。即使是从不同供应商处购买的相同辅料,其亚硝酸盐浓度也可能截然不同,这可能是由于生产工艺的差异所致6。
该方法可处理的样品基质种类更多、样品检测量更大,因此需要开发一种常规方法来评估辅料中的亚硝酸盐浓度,让制药生产商确信所用原料无潜在污染 - 不受供应商、辅料类型、甚至使用批次的影响。此类方法应考虑到标记受污染的原材料,以便必要时可以进一步确认检测。这种风险评估应快速且经济高效,尽管低成本方法常用于亚硝酸盐评估,但这些方法通常耗时且灵敏度较差,并且易受干扰7。
液相色谱与单四极杆质谱联用(LC-MS)方法可提供亚硝酸盐痕量分析所需的灵敏度和选择性,但归根结底,任何解决方案还必须具备高通量常规工作流程所需的稳健性和可靠性,同时保持每个样品的低分析成本8。
在下文所述的实验中,采用了UHPLC联用单四极杆质谱检测器,来评估这种分析配置在29天的分析过程中对制药辅料进行长期分析的适用性。本研究的目的是展示ACQUITY QDa II质谱检测器随时间推移的可靠性和稳健性。研究中采用的方法源自Jireš和Douša的UPLC™方法,具有分析速度快、灵敏度高和检测选择性强的优势8。 本分析将现有方法转换至ACQUITY Arc™系统,这是一种经济高效的替代方法,同时仍具备有效分析制药辅料所需的性能、稳健性和可靠性。
实验
购买市售的辅料样品并在室温下保存,直至使用。选择山梨糖醇、乳糖一水合物、玉米淀粉、麦芽糖糊精和碳酸钙作为典型制药辅料的代表。所有购买的化学品和试剂均为分析纯质量,所有样品、溶液和流动相前准备均采用UPLC级溶剂。
样品萃取遵循Jireš和Douša修改后的方案(如图1所示),其中粉末状辅料通过2,3-二氨基萘(DAN)衍生化方法处理8。 该方法的原理是亚硝酸根离子与DAN反应形成1H-萘并(2,3-d)三唑(NAT),然后使用质谱检测器进行分离和检测。随后定量分析NAT化合物,作为样品中总亚硝酸根生成的替代指标。
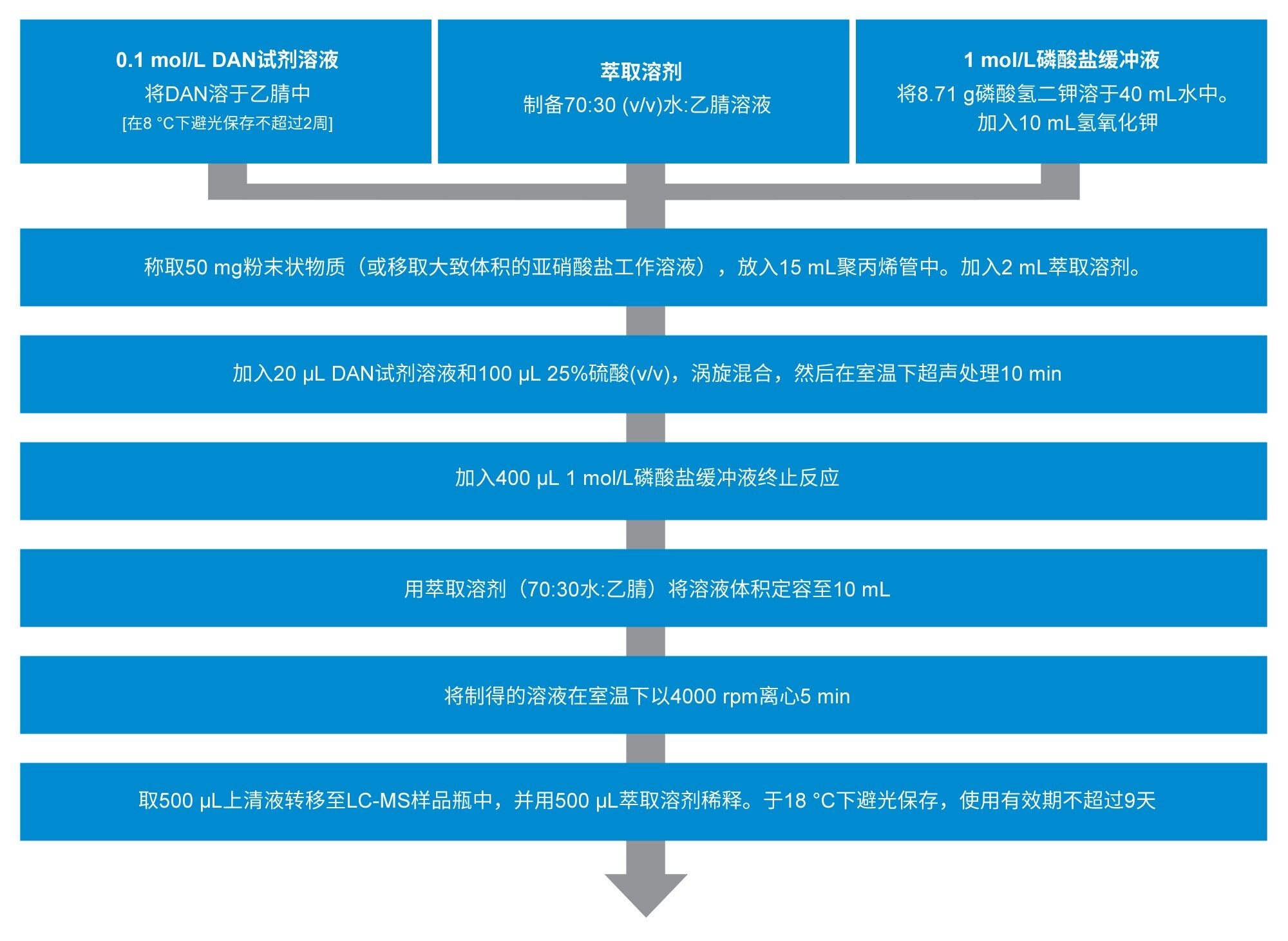 图1.样品前处理采用Jireš和Douša (2022)的方法。
图1.样品前处理采用Jireš和Douša (2022)的方法。
使用亚硝酸盐加标溶液(1 µg/mL)构建标准曲线,并将该溶液以不同浓度添加到空聚丙烯管中(0.5、5.0和12.5 ng/mL,分别对应辅料中的0.2、2.0和5.0 µg/mL),并相应地通过样品前处理程序处理。然后根据标准曲线对未加标的辅料样品进行定量。此外,我们还制备了一份过程空白样(程序空白样品),用以评估样品前处理过程中固有的亚硝酸盐相互作用基线水平。过程空白样中使用100 µL萃取溶剂代替加标溶液。
在每种辅料类型的50 mg样品中加标相应量的溶液(浓度为0.5、5.0和12.5 ng/mL),来评估回收率。每个批次的所有样品前处理均重复三次。
为了在稳健性测试中评估并监测系统稳定性,我们使用萃取溶剂制备了两种浓度水平(0.5 µg/mL和5 µg/mL)的含有对乙酰氨基酚和咖啡因的仪器QC标准品,并在整个分析过程中执行分析。
样品前处理工作按照所述样品前处理技术进行,共分为四个连续的样品批次,这意味着整个样品前处理过程总共重复了四次。选择离子监测(SIR)使用目标离子进行采集,分别为:m/z 170.1作为NAT的[M+H]+,m/z 152.1作为对乙酰氨基酚的[M+H]+,以及m/z 195.1作为咖啡因的[M+H]+。
分析采用ACQUITY Arc系统联用ACQUITY QDa II质谱检测器。为了确保Jireš和Douša方法中规定的UPLC条件能够在不超出系统压力限制的情况下采用并转移到UHPLC系统,研究中使用了方法转换计算器2.0版软件(667005222)8。 这样用户可以在不影响系统预期性能和灵敏度的前提下,使用规格较低的LC系统,从而确保实现经济高效的分析。
LC-MS实验条件
|
LC系统: |
配备FTN-R样品管理器的ACQUITY Arc |
|
检测: |
ACQUITY QDa II质谱检测器 |
|
色谱柱: |
ACQUITY UPLC HSS™ T3色谱柱, 100 Å, 1.8 µm, 2.1 mm × 100 mm(P/N:186003539) |
|
柱温: |
45 ℃ |
|
样品温度: |
18 °C |
|
进样体积: |
5 µL |
|
流速: |
0.4 mL/min |
|
运行时间: |
6 min |
|
流动相A: |
0.1%甲酸水溶液 |
|
流动相B: |
乙腈 |
|
样品瓶: |
12 × 32 mm透明玻璃螺纹颈口样品瓶,100个/包(P/N:186000273) |
|
电离模式: |
正离子电喷雾(ES+) |
|
毛细管电压: |
0.8 kV |
|
脱溶剂气温度: |
400 °C |
|
离子源温度: |
120 °C |
|
采集模式: |
SIR |
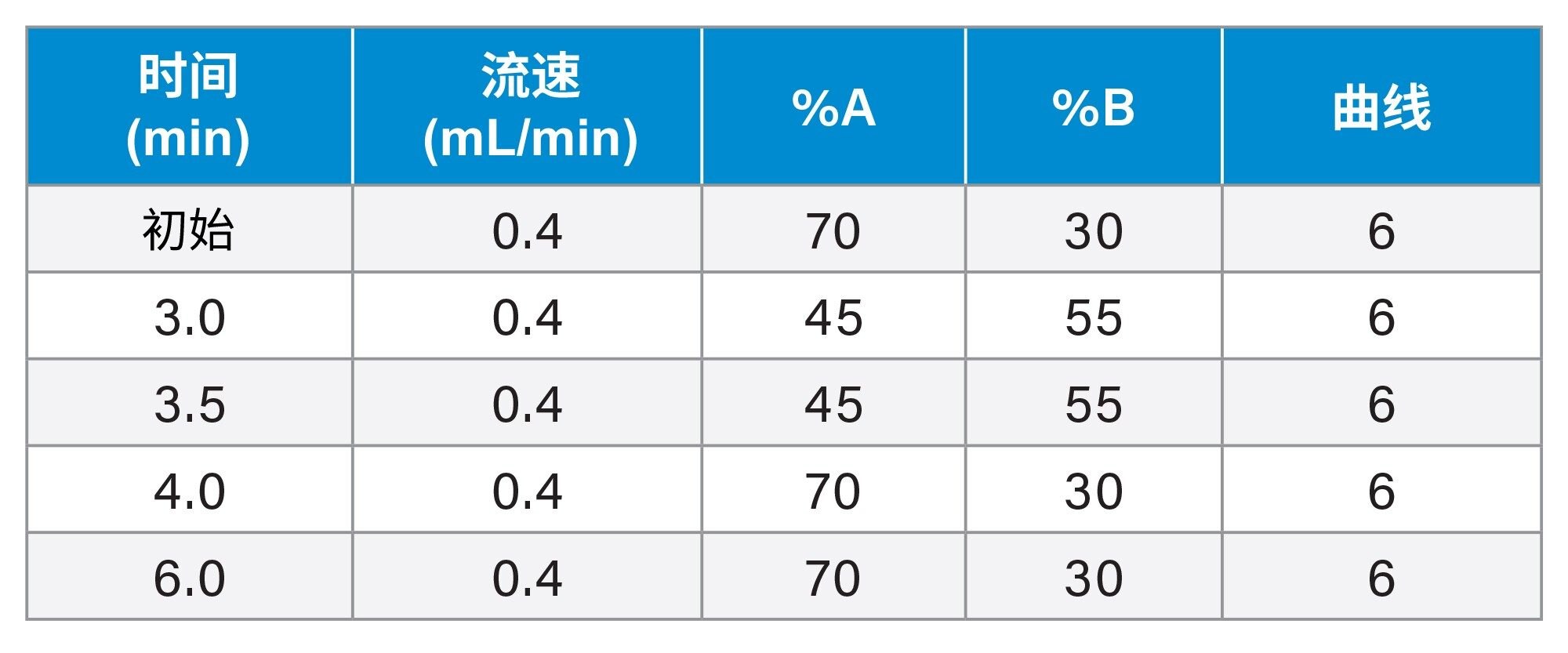
LC梯度
软件
|
数据采集、处理和报告: |
Empower™ CDS 3.0 |
结果与讨论
方法性能
使用上述LC-MS方法条件,通过定量分析NAT (m/z 170),实现了对所选辅料中亚硝酸盐含量的快速测定。NAT化合物的保留时间(RT)约为2.73 min。样品通过标准曲线进行定量,定量范围为0.5~12.5 ng/mL,对应于辅料样品中0.2~5.0 µg/g的亚硝酸盐浓度(µg/g = ppm)。
该方法的LOQ受限于工艺空白样中存在的亚硝酸盐。图2展示了亚硝酸盐标准品的绝对峰面积与过程空白的对比相关示例。
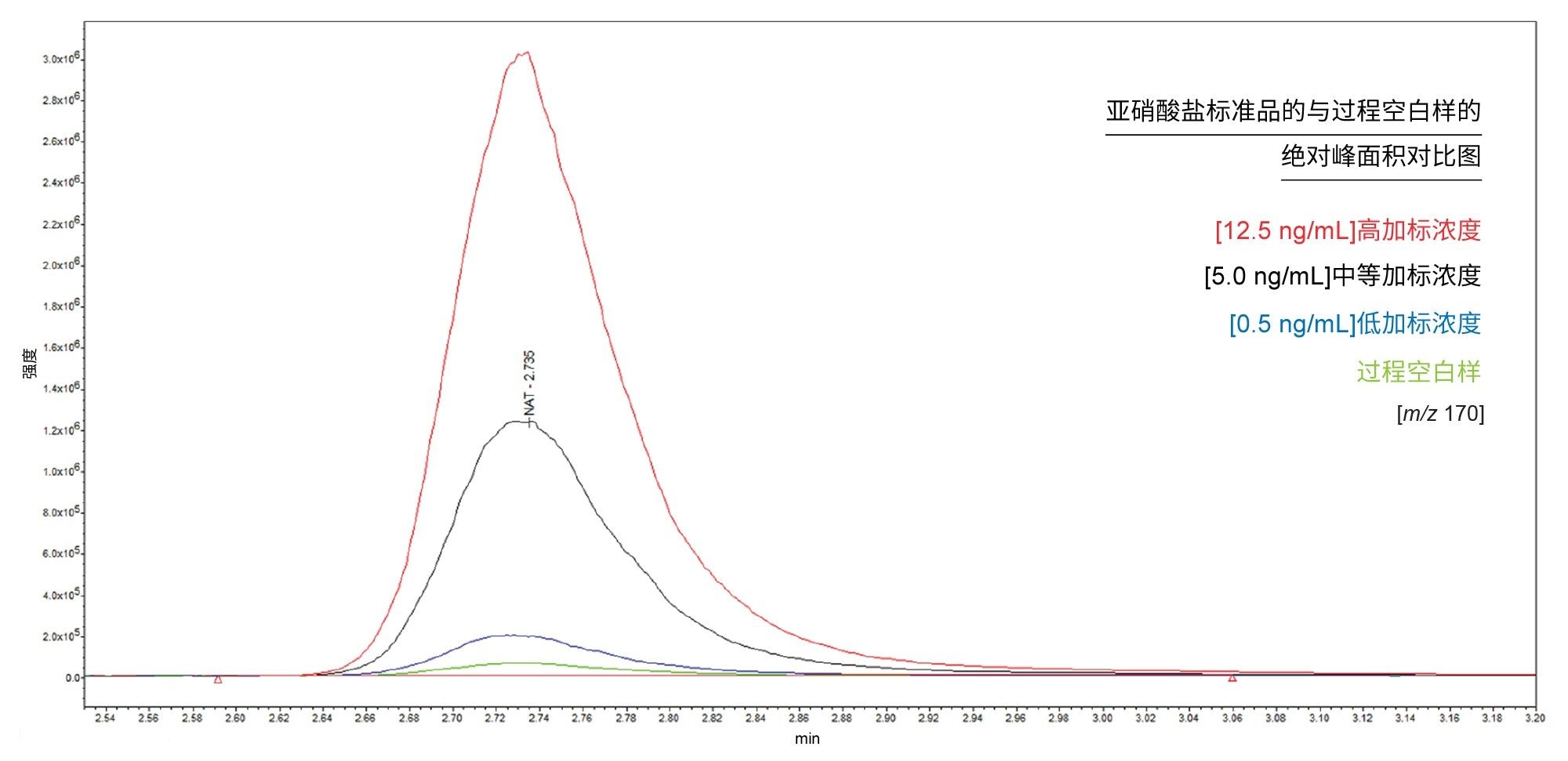 图2.给定重复样品序列亚硝酸盐标准品与过程空白样的绝对峰面积叠加对比图。
图2.给定重复样品序列亚硝酸盐标准品与过程空白样的绝对峰面积叠加对比图。
标准曲线图是使用修正峰面积构建的,其中过程空白的峰面积平均值(每组样品中n=6)被求和,然后从所有标准品和未知样品的峰面积中减去该值,公式如下:修正峰面积 = 峰面积 – (BL?.%..AVE(峰面积))。使用这种空白扣除方法,经过调整后的标准曲线图的X轴截距就成为了空白样品的代表值,即每个样品中由于样品前处理技术的污染而存在的亚硝酸盐水平。然后根据截距以上的响应增量来定量后续样品中的亚硝酸盐含量。
为了代表高通量工作流程,我们创建了一个预定义的进样序列(181次进样),并在148~190 h的样品批次中多次分析该序列。此处的每个样品批次(n = 4)表示制备新样品 - 包括加标和未加标辅料、亚硝酸盐标准品、过程空白和QC样品。样品前处理在总共29天的分析过程中重复四次,共执行32次进样序列的重复。该分析旨在展示系统的稳健性,设计为持续无中断分析,除了例行更换LC流动相和溶剂。任何额外的操作(例如离子源清洁)均记录在表1中。
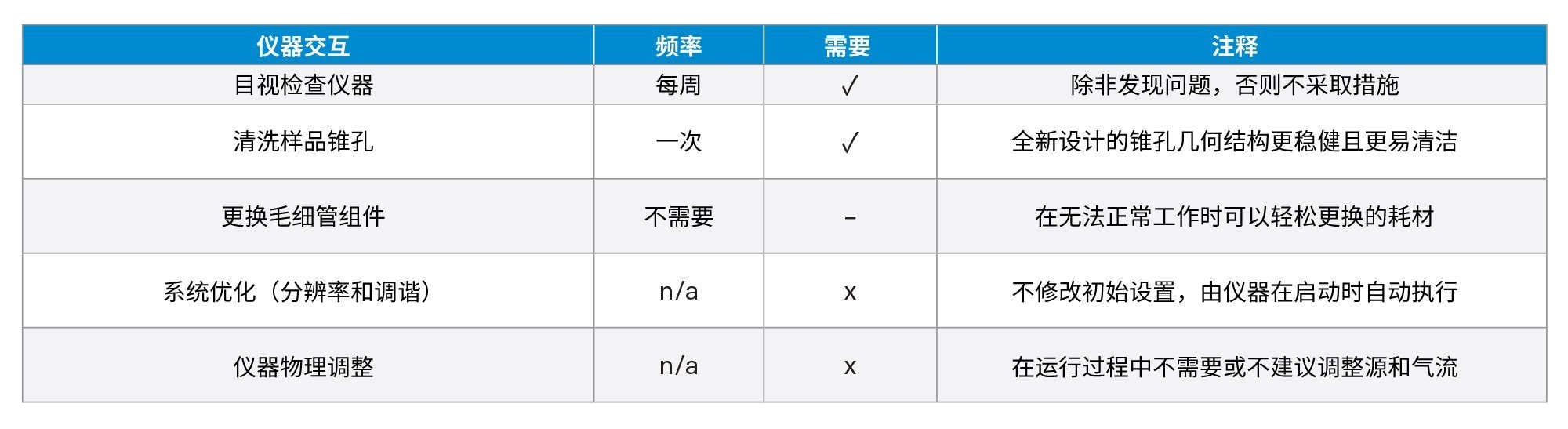 表1.ACQUITY QDa II质谱检测器的典型仪器交互,以及分析期间每个任务的执行频率
表1.ACQUITY QDa II质谱检测器的典型仪器交互,以及分析期间每个任务的执行频率
在整个分析过程中,针对预定义的进样序列(n=32)的每个重复样品,都在Empower中绘制了标准曲线。每个进样序列均单独处理,并包含间插标准曲线标准品的辅料样品,所有样品均重复制备三份。除批次2的标准曲线(R2 > 0.97)外,其余所有进样序列的标准曲线均为线性(1/X),且R2 >0.99。图3展示了给定样品批次的第一个和最后一个标准曲线图的比较,两者之间的分析时间间隔接近150 h。在本例中,任何给定数据点对应的残差均不超过3.7%。
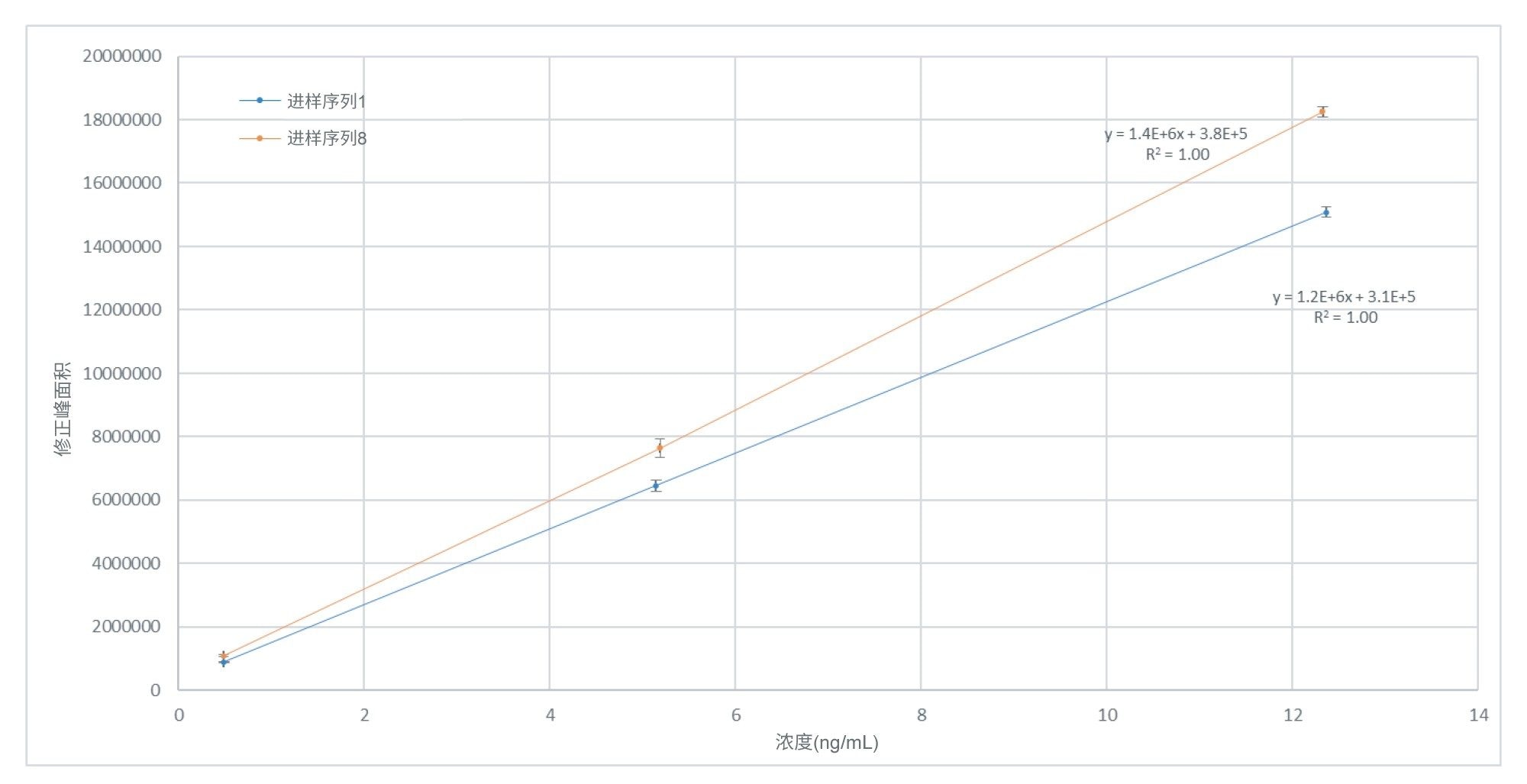 图3.亚硝酸盐(以NAT计算)的标准曲线响应在0.5~12.5 ng/mL(相当于原材料中0.2~5 µg/g的亚硝酸盐)浓度范围内进行分析,标准曲线标准品在样品批次开始和结束时重复进样品进行了三次进样分析。每个标准曲线水平的平均值及其相应的标准偏差基于n=6次进样。
图3.亚硝酸盐(以NAT计算)的标准曲线响应在0.5~12.5 ng/mL(相当于原材料中0.2~5 µg/g的亚硝酸盐)浓度范围内进行分析,标准曲线标准品在样品批次开始和结束时重复进样品进行了三次进样分析。每个标准曲线水平的平均值及其相应的标准偏差基于n=6次进样。
与之前的样品批次相比,分析最终样品批次的时间略长(190 h),因此最适合展示方法稳定性随时间的变化。考虑到本研究的目的,通过比较加标辅料的峰面积与已知等效加标水平的峰面积来评估回收率。图4展示了最终样品批次的回收率,范围为70%~129%,其中唯一不在可接受回收范围内的是0.5 ng/mL(0.2 µg/g)加标的玉米淀粉。所有加标样品的总平均回收率为94%。
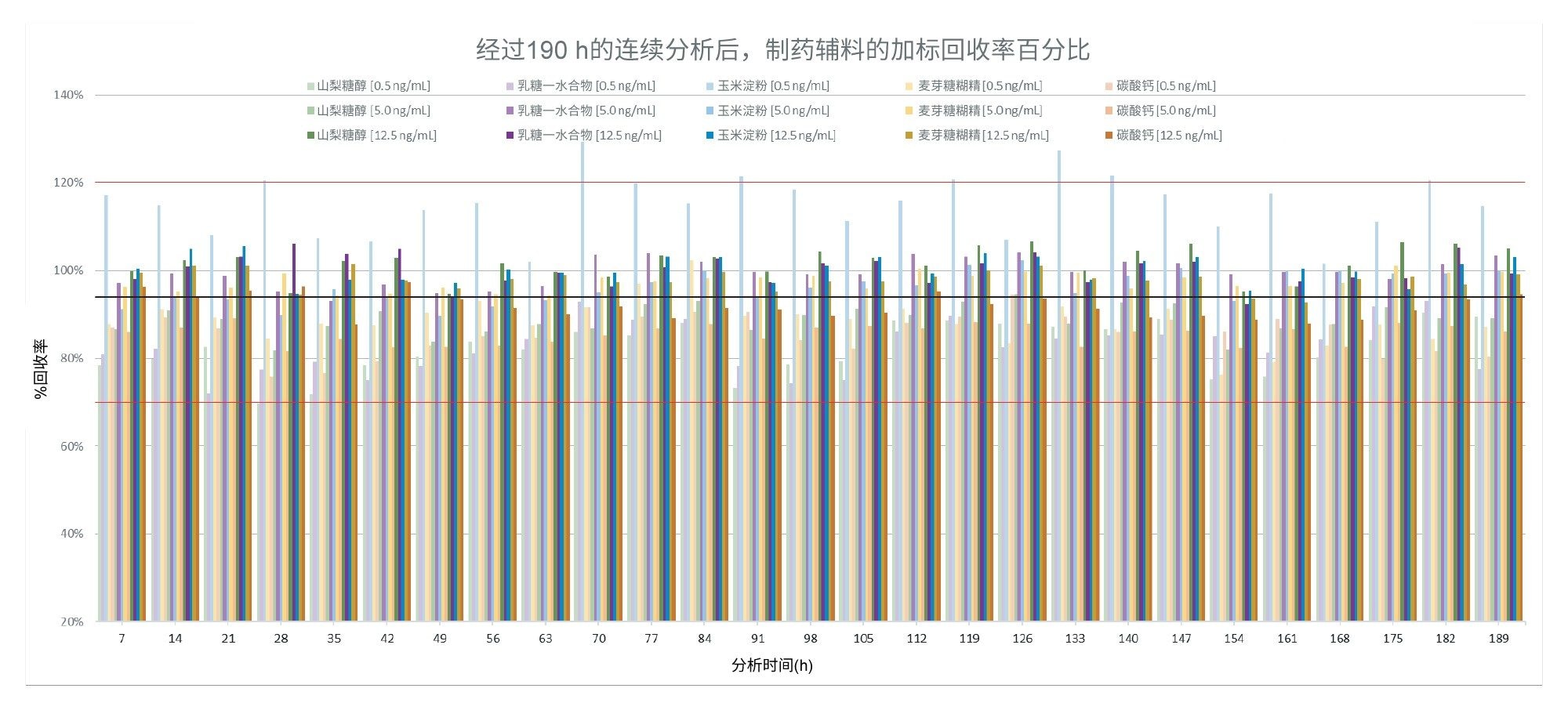 图4.在190小时的连续采集过程中,所有加标辅料样品的亚硝酸盐回收率(%)。红线表示70%~120%的容差区间,黑线表示94%的总平均回收率。
图4.在190小时的连续采集过程中,所有加标辅料样品的亚硝酸盐回收率(%)。红线表示70%~120%的容差区间,黑线表示94%的总平均回收率。
图5显示了给定序列中每种未加标辅料的绝对峰面积的代表性示例。重要的是,过程空白样和辅料样品使用相同的样品前处理技术,因此可以假设过程空白样中存在的固有水平的污染也可能出现在所有辅料样品中。在图5中观察到的过程空白样品中的NAT浓度为0.041 ng/mL。因此,该图展示了在分析的所有辅料中(玉米淀粉除外),亚硝酸盐的相对含量不高。
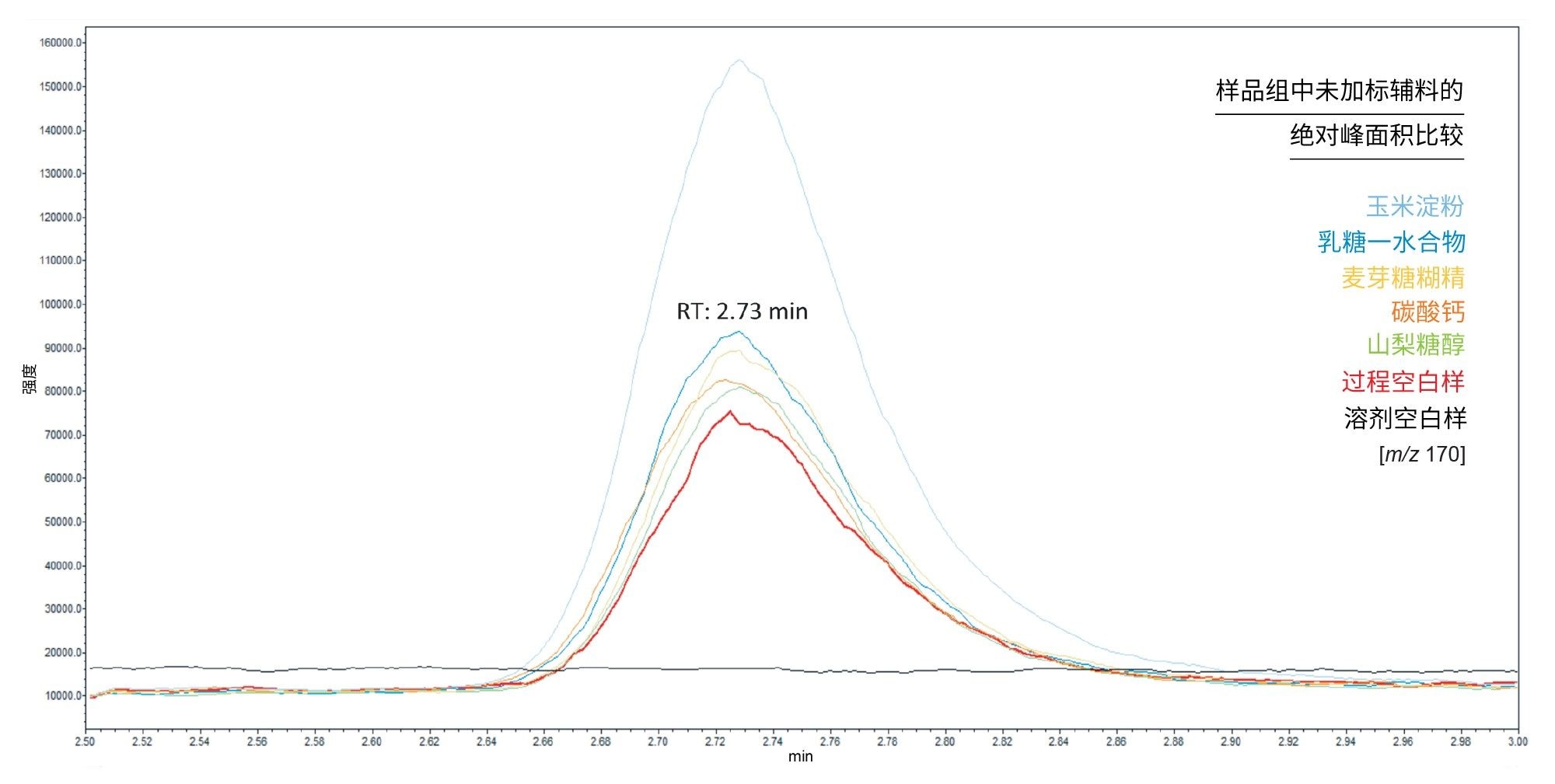 图5.给定样品序列中的过程空白样品与未加标辅料样品相比,亚硝酸盐绝对峰面积的SIR叠加色谱图。
图5.给定样品序列中的过程空白样品与未加标辅料样品相比,亚硝酸盐绝对峰面积的SIR叠加色谱图。
在分析过程中,限制因素是过程空白样中存在的亚硝酸盐,而不是MS灵敏度。这种亚硝酸盐的存在源于样品前处理过程中各种来源的污染。该方法本身具有良好的重现性,在采集的四个样品批次中,过程空白样计算值的%RSD范围为2.7%~8.5%,但未来的工作应着重于消除这种污染上,以降低方法的检测限(LOD))。
在分析过程中对五种辅料进行了检测,结果显示只有玉米淀粉的亚硝酸盐含量可能需要进一步分析。图6展示了经过190 h分析后得到的未加标玉米淀粉样品的浓度报告值,其亚硝酸盐含量的平均值为0.14 µg/g,在n=27次进样中的%RSD为8.7%。该值与辅料数据库给出的玉米淀粉的平均值一致6。 虚线表示玉米淀粉标示报告阈值的假设示例,该阈值绘制为文献中报告的亚硝酸盐含量平均值的0.5倍。在本示例中,辅料的亚硝酸盐含量超过了该阈值,因此会被标记为需要进一步分析。
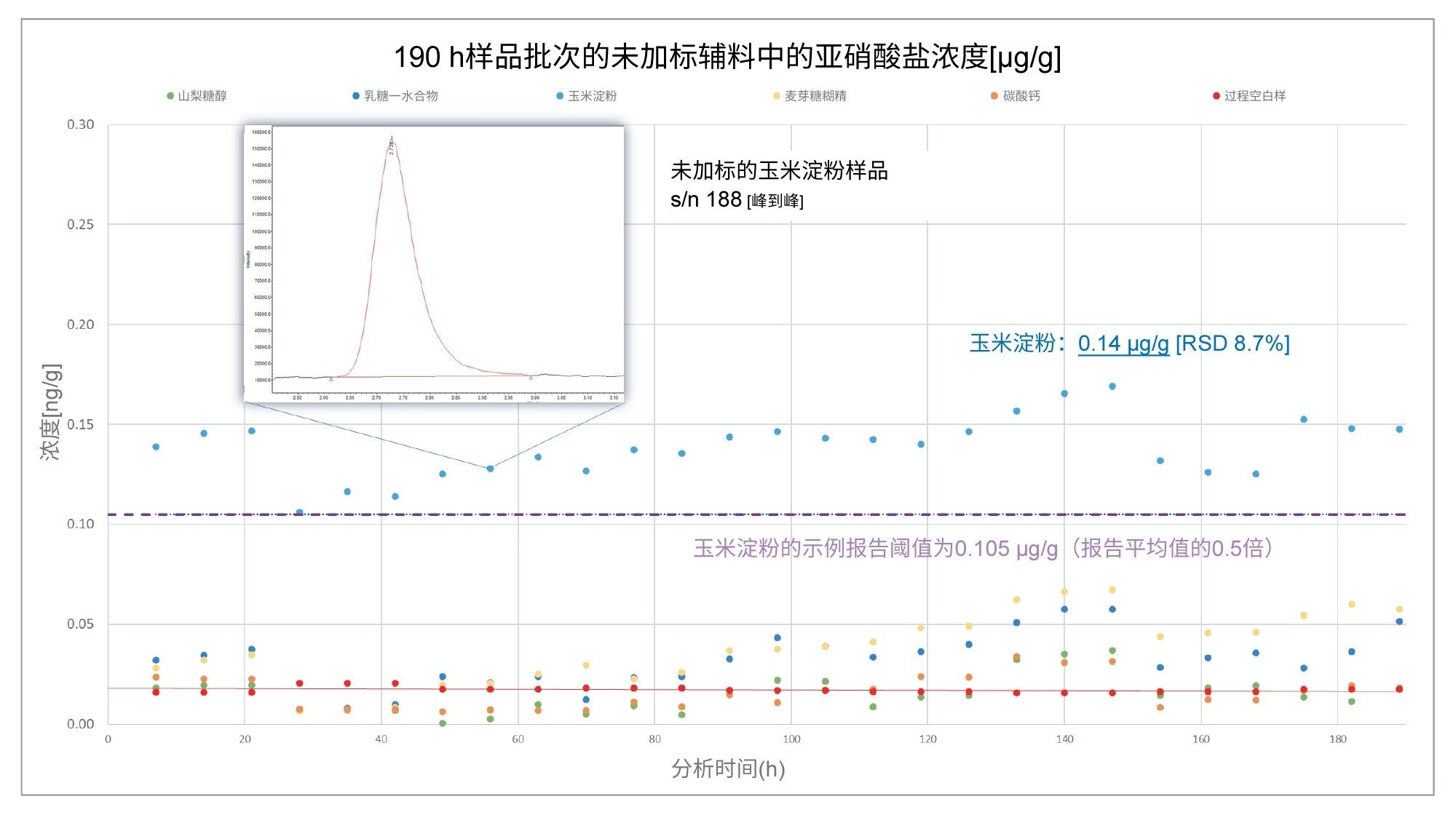 图6.在190 h的连续采集内分析未加标的辅料样品中的亚硝酸盐含量(µg/g)。玉米淀粉样品中的亚硝酸盐含量超过了文献报道的0.5倍平均值0.105 µg/g。对该样品进行n=27次进样的亚硝酸盐测量精密度显示的%RSD为8.7%。
图6.在190 h的连续采集内分析未加标的辅料样品中的亚硝酸盐含量(µg/g)。玉米淀粉样品中的亚硝酸盐含量超过了文献报道的0.5倍平均值0.105 µg/g。对该样品进行n=27次进样的亚硝酸盐测量精密度显示的%RSD为8.7%。
相比之下,在图7中,使用相同的处理确定乳糖一水合物的示例报告阈值,分析的辅料未超过0.27 µg/g的阈值,因此在这种情况下,可以认为它是合格的,可以使用。
这里%RSD的较大变化反映了辅料中的亚硝酸盐含量与过程空白样非常接近。两者之间浓度的相对差异非常小,表明辅料中未检出亚硝酸盐。因此,高%RSD值反映的是方法局限性,而非仪器性能问题。
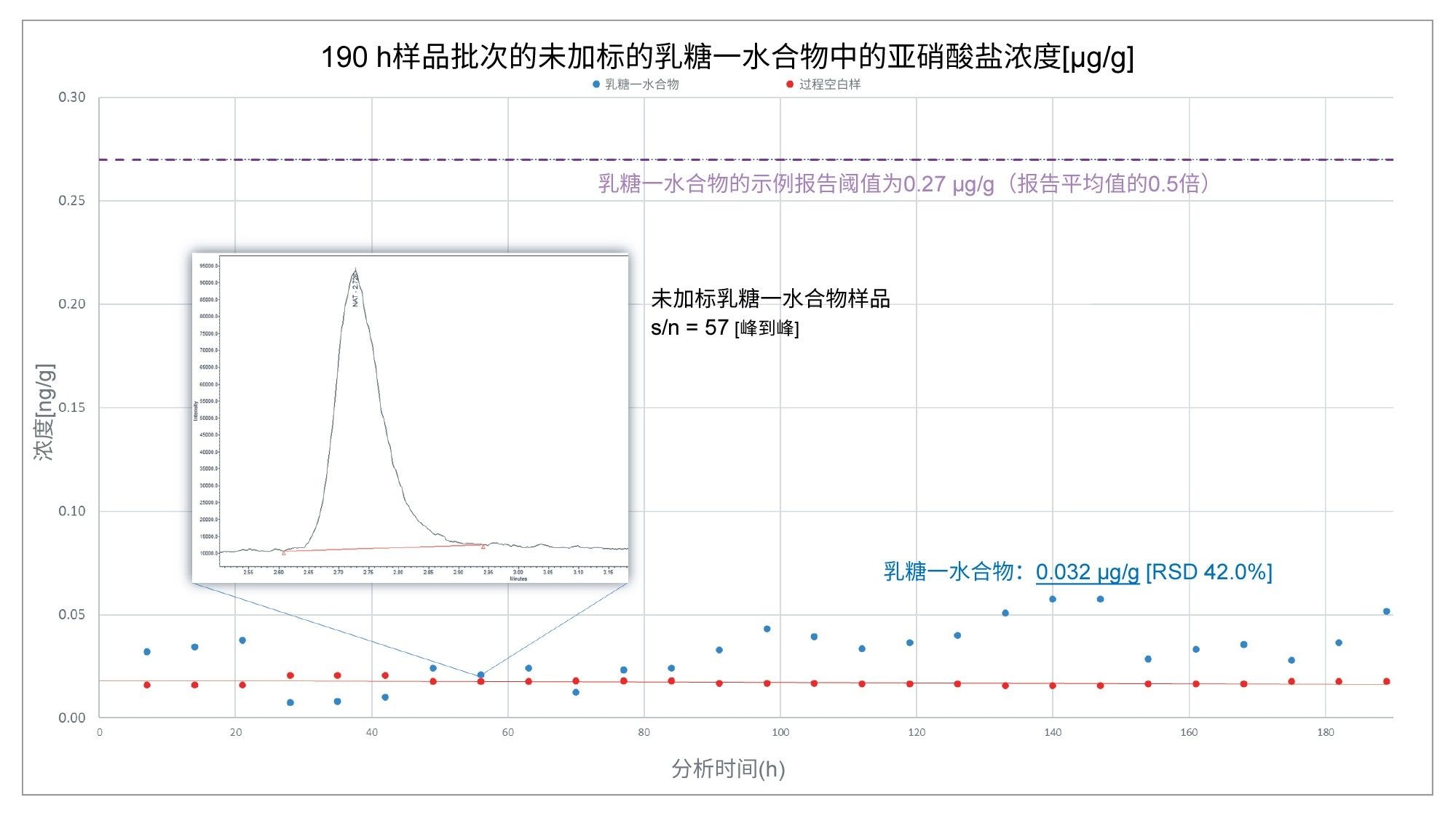 图7.在190小时的连续分析过程中,未加标的乳糖一水合物的亚硝酸盐含量(µg/g)。乳糖一水合物样品中的亚硝酸盐含量没有超过文献报道的0.5倍平均值0.27 µg/g。
图7.在190小时的连续分析过程中,未加标的乳糖一水合物的亚硝酸盐含量(µg/g)。乳糖一水合物样品中的亚硝酸盐含量没有超过文献报道的0.5倍平均值0.27 µg/g。
图6中还绘制了给定样品批次的其他未加标辅料的值。这些数值也没有显著超过工艺空白的阈值,无法准确定量。同样,这也表明这些辅料中不存在亚硝酸盐,或者亚硝酸盐的水平低至方法检测限,因此这些样品将不会标记为需要进一步分析。
在图7中,未加标的乳糖一水合物样品的信噪比表明,方法的灵敏度受限于过程空白样,而不是仪器性能。系统的灵敏度(如上图中嵌入的示例色谱图所示)足以检测略高于(图6)和远低于(图7)报告阈值的浓度。之前报告的数据表明,该方法的LOD为0.0013 ng/mL8。
使用Empower CDS报告功能可以轻松查看样品批次中辅料的亚硝酸盐浓度,如图8所示。这有助于快速制定全面的决策,同时确保实验室的合规性和数据可靠性。在所示的示例中,未加标的玉米淀粉样品的报告浓度为0.392 ng/mL(或0.157 µg/g)。
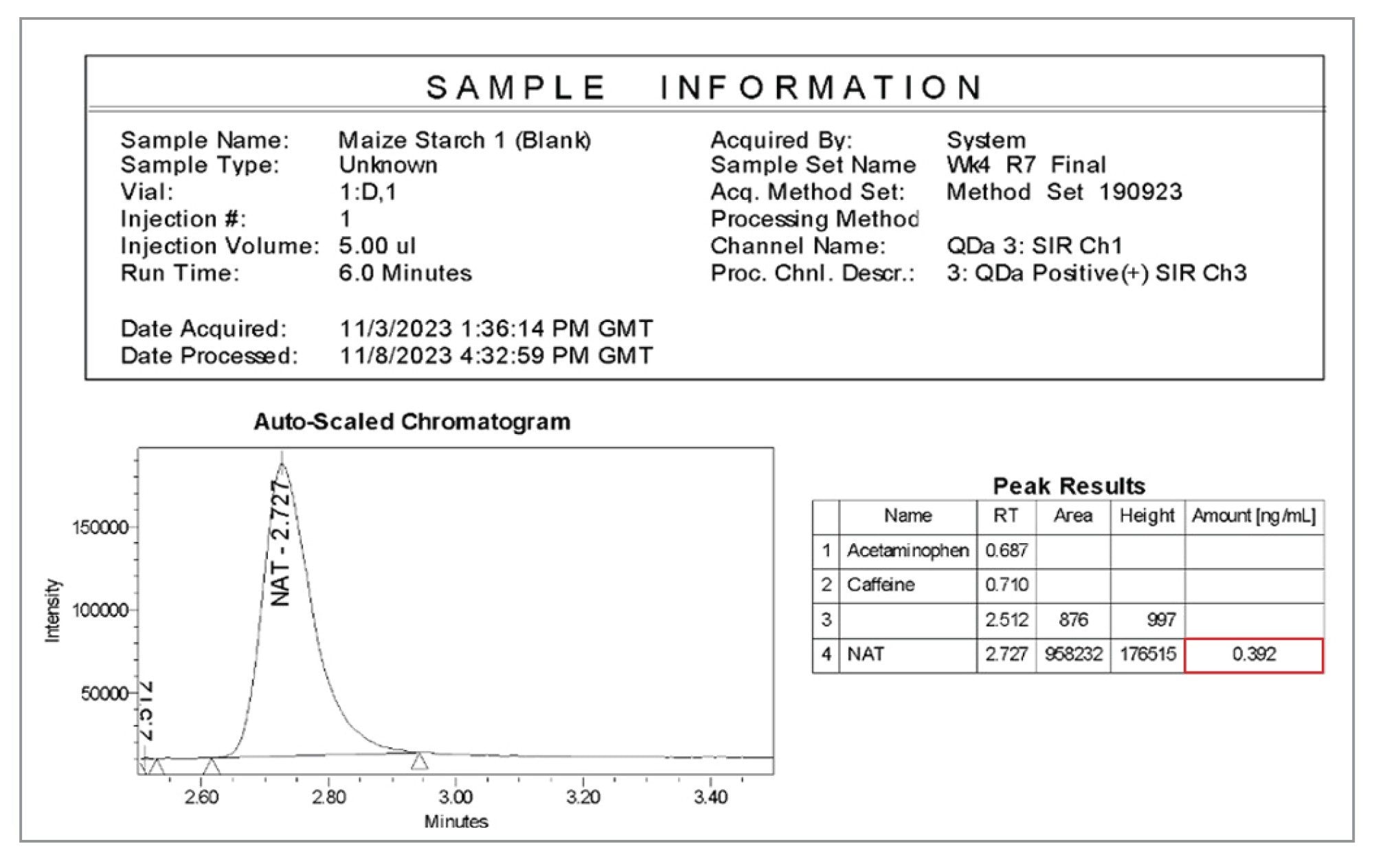 图8.Empower CDS生成的玉米淀粉样品中亚硝酸盐含量数据的报告示例。
图8.Empower CDS生成的玉米淀粉样品中亚硝酸盐含量数据的报告示例。
仪器稳健性
除方法性能外,研究中还评估了仪器的总体正常运行时间和可靠性,并记录了分析过程中的仪器交互和所有维护程序。为期29天的实验中,我们使用该仪器总共执行了5792次进样,其中1920次进样为辅料样品。分析概览如图9所示。
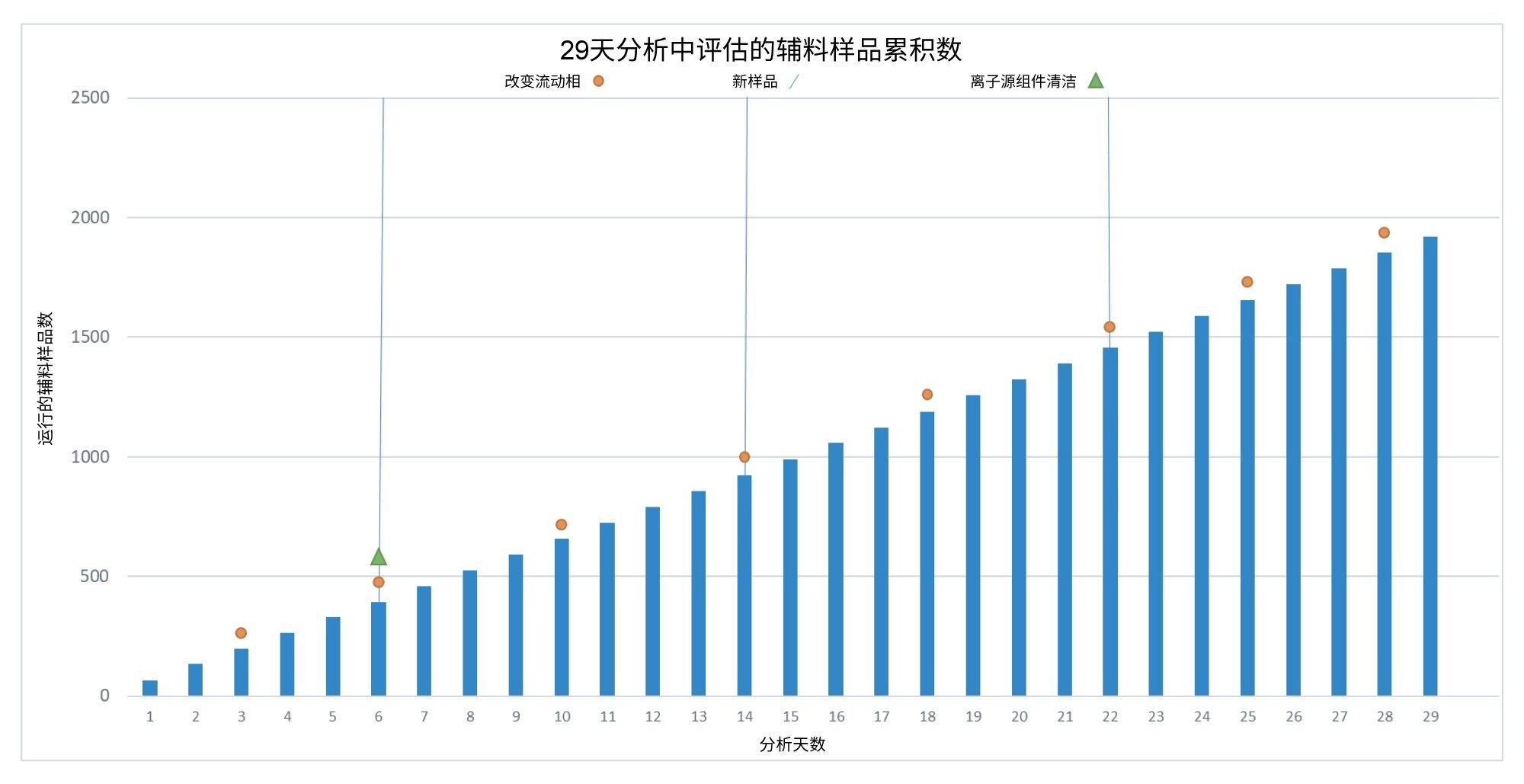 图9. 29天分析中辅料样品的累积图 - 展示了流动相的变化、新样品的引入,以及所需的任何清洁操作。
图9. 29天分析中辅料样品的累积图 - 展示了流动相的变化、新样品的引入,以及所需的任何清洁操作。
在29天的分析期间,系统交互非常少。在676小时的批量分析过程中,仪器仅需要两个小时的维护,包括20分钟的排气和清洁过程。系统在两小时内恢复分析。尽管在6天后清洁了离子源,但系统在随后的23天中保持全面运行,这表明污染可能与之前的仪器使用有关。其他形式的仪器交互详细信息见表1。
由于QC进样的结果超出了可接受范围,因此对离子源组件进行了一次清洗。仪器分离度、调谐和校正在启动过程中自动完成,无需分析人员干预。
流动相和清洗溶液根据需要补充。在任何溶剂空白(整个分析中n=1920)中均未观察到亚硝酸盐残留,表明此方法有望实现更高的样品通量。
QDa II质谱检测器的稳定性是评估该工作流程可行性的关键指标。 在分析过程中观察了仪器QC样品(每个浓度n=768个),以独立于亚硝酸盐样品单独评估仪器的稳健性。整个分析的结果一致。在任何给定的样品批次中,给定QC水平的%RSD均未超过9.8%。在清洁离子源组件后520 h的连续运行条件下,QC化合物和加标水平的%RSD均小于12.5%,如图10所示。
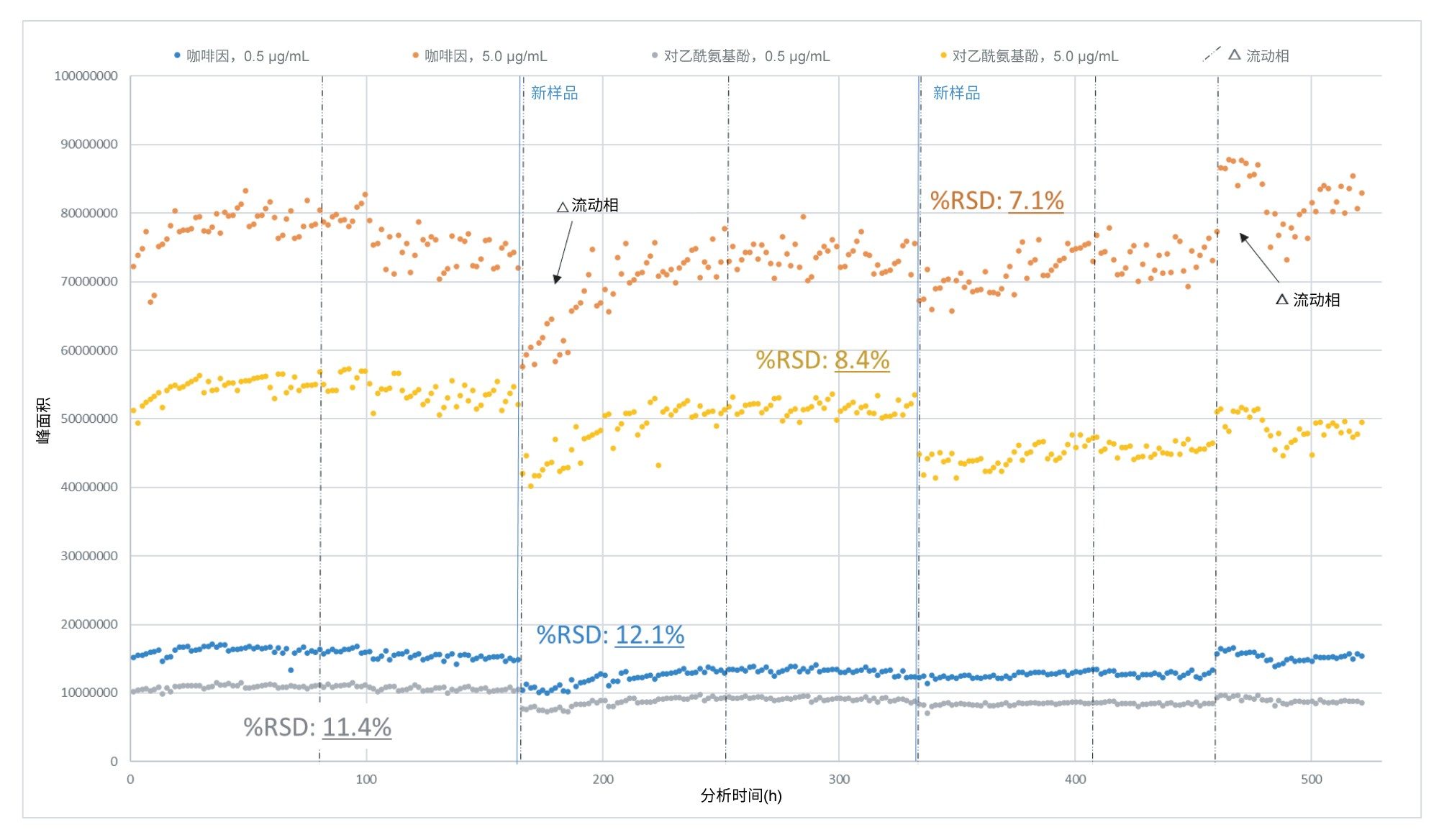 图10.在520 h的连续分析中,QC组分(咖啡因和对乙酰氨基酚)的绝对峰面积(排除样品锥孔清洗之前的数据点)。
图10.在520 h的连续分析中,QC组分(咖啡因和对乙酰氨基酚)的绝对峰面积(排除样品锥孔清洗之前的数据点)。
通过评估亚硝酸盐标准品进样的绝对峰面积%RSD,也可以证明每个样品批次中仪器的稳健性。在整个分析过程中,任何给定样品批次的%RSD均未超过14.7%,如表2所示。
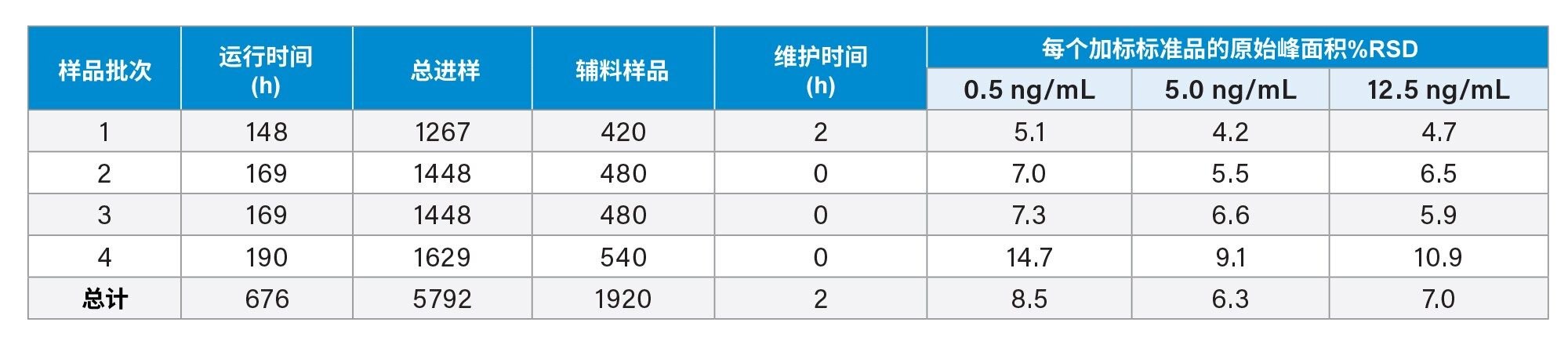 表2.稳健性研究过程中分析的进样次数和辅料样品明细,以及加标标准品的绝对峰面积%RSD(对于任何给定的加标水平,n≥42)。
表2.稳健性研究过程中分析的进样次数和辅料样品明细,以及加标标准品的绝对峰面积%RSD(对于任何给定的加标水平,n≥42)。
报告的各辅料加标浓度大多符合预期。在整个分析过程中,所有加标水平和辅料类型的%RSD均低于20%。示例见图11,其中显示了整个分析中所有加标水平的碳酸钙的%RSD均不超过13.9%。
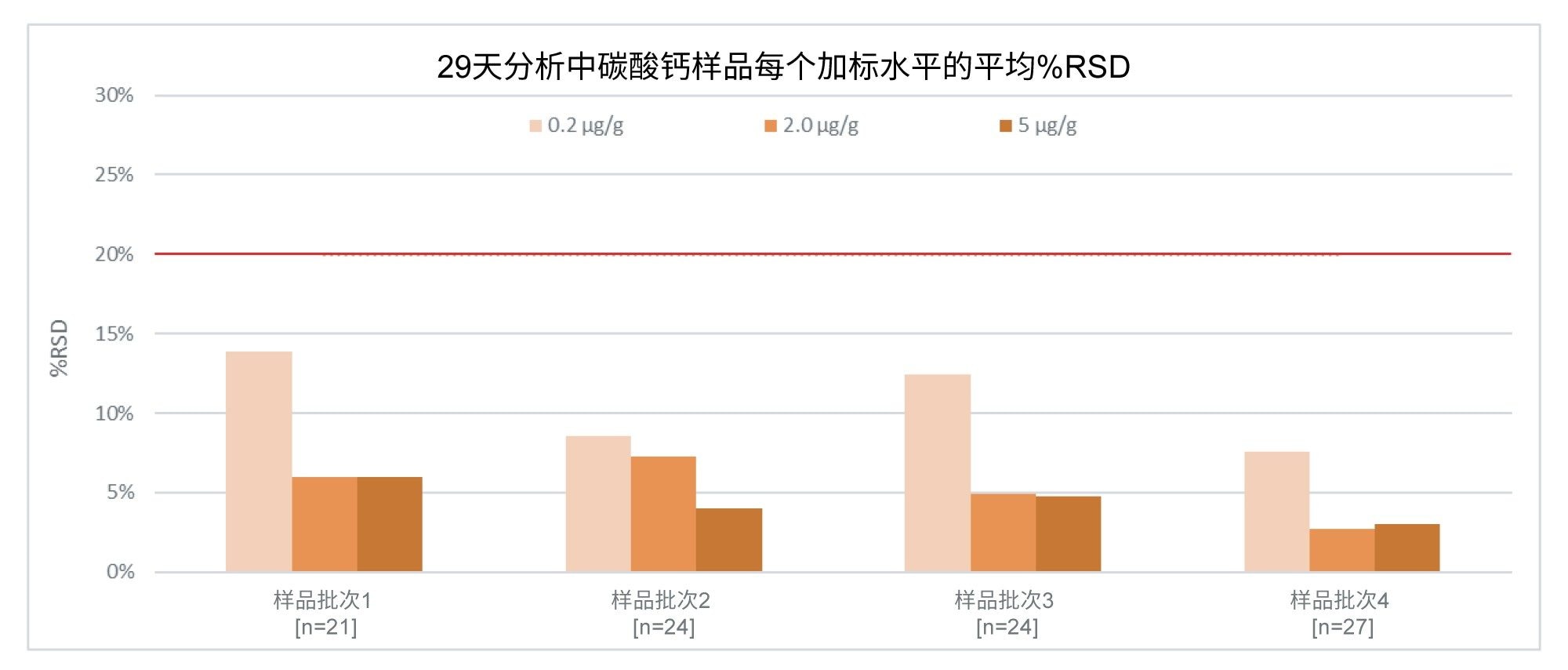 图11.碳酸钙辅料样品中0.2、2.0和5.0 µg/g亚硝酸盐加标的平均%RSD值,基于每个加标水平在给定样品批次中的(n)次重复测定。红线表示%RSD的阈值20%。
图11.碳酸钙辅料样品中0.2、2.0和5.0 µg/g亚硝酸盐加标的平均%RSD值,基于每个加标水平在给定样品批次中的(n)次重复测定。红线表示%RSD的阈值20%。
图12展示了在29天的分析过程中,每个辅料样品和加标水平的平均%RSD值。在整个分析过程中,五种辅料在0.2、2.0和5.0 µg/g (ppm)加标水平的%RSD分别未超过18.5%、11.1%和6.0%,展示了仪器性能随时间的稳健性。
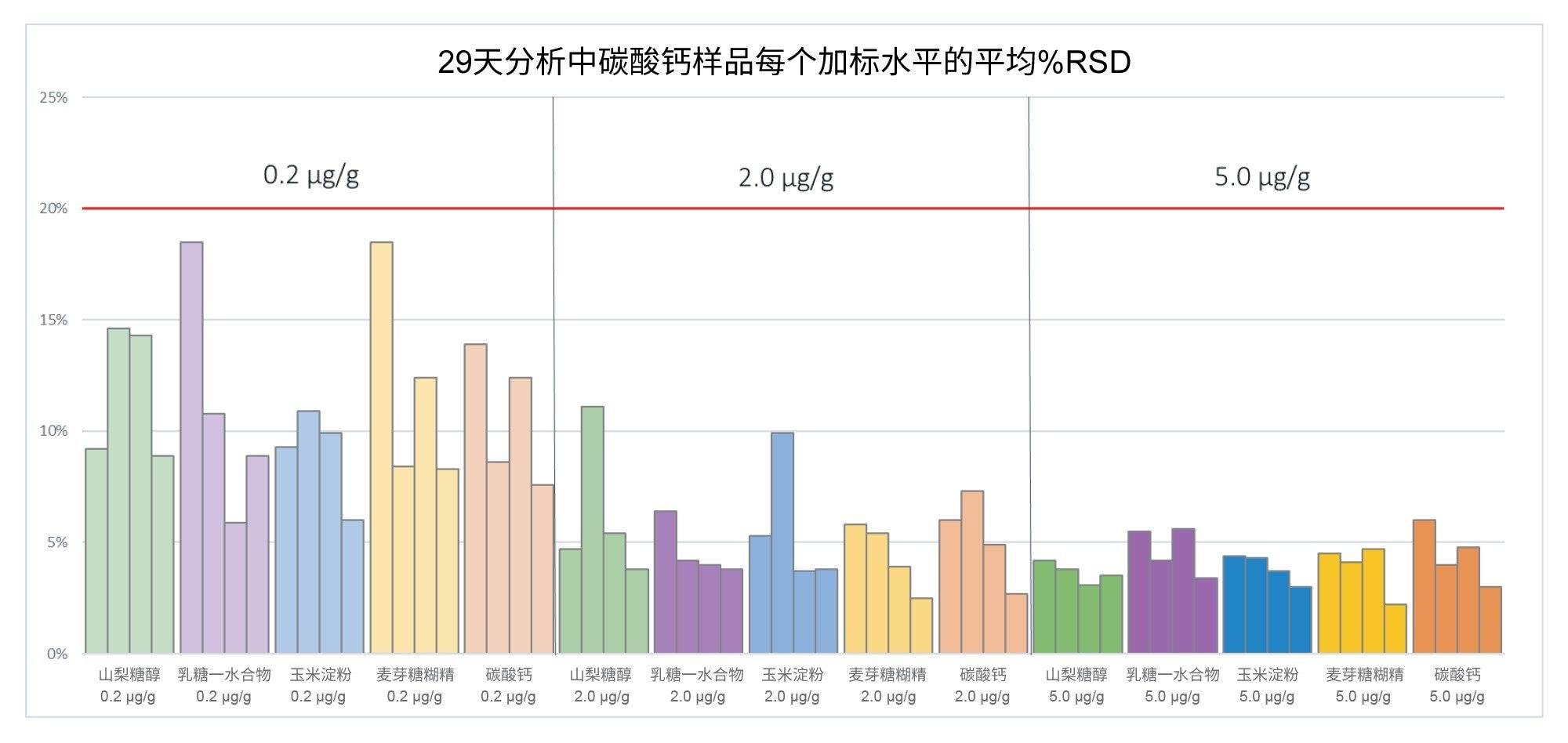 图12.整个分析过程中所有辅料加标水平的平均%RSD,其中每个辅料加标水平的四个数据点分别代表给定样品批次中某一加标水平的平均值(注意:由于样品前处理错误,样品批次1中0.2 µg/g麦芽糊精加标水平的一个重复样品被排除)。
图12.整个分析过程中所有辅料加标水平的平均%RSD,其中每个辅料加标水平的四个数据点分别代表给定样品批次中某一加标水平的平均值(注意:由于样品前处理错误,样品批次1中0.2 µg/g麦芽糊精加标水平的一个重复样品被排除)。
结论
- 实验结果表明,ACQUITY QDa II质谱检测器是一款稳健可靠的仪器,适用于常规的高通量应用,如制药辅料中亚硝酸盐的分析
- 在676小时的分析过程中,该仪器成功分析了约6000次进样,停机时间仅为2小时
- 给定样品批次中,仪器QC进样的%RSD不超过9.8%,表现出一致的性能
- 在长达520小时的连续分析中,任何给定QC水平的绝对峰面积%RSD均低于12.5%,表明系统在整个分析过程中具有出色的稳定性
- 在任何给定的样品批次分析中,所有亚硝酸盐标准品进样的绝对峰面积%RSD始终小于14.7%
- 在玉米淀粉辅料中成功检测到0.14 µg/g的亚硝酸盐浓度,与辅料数据库中报告的平均浓度一致,表明该方法具有良好的定量准确性。其他辅料在分析过程中未展示出可检出水平的亚硝酸盐
- 该方法使用ACQUITY Arc UHPLC和ACQUITY QDa II质谱检测器,运行时间6 min,是一种高效且经济的方法,适用于在制药辅料分析中定量µg/g级亚硝酸盐
参考资料
- Brambilla, G., & Martelli, A. (2007).Genotoxic and carcinogenic risk to humans of drug-nitrite interaction products.In Mutation Research - Reviews in Mutation Research (Vol.635, Issue 1, pp.17–52). https://doi.org/10.1016/j.mrrev.2006.09.003.
- Horne, S., Vera, M. D., Nagavelli, L. R., Sayeed, V. A., Heckman, L., Johnson, D., Berger, D., Yip, Y. Y., Krahn, C. L., Sizukusa, L. O., Rocha, N. F. M., Bream, R. N., Ludwig, J., Keire, D. A., & Condran, G. (2023).Regulatory Experiences with Root Causes and Risk Factors for Nitrosamine Impurities in Pharmaceuticals.In Journal of Pharmaceutical Sciences (Vol.112, Issue 5, pp.1166-1182).Elsevier B.V. https://doi.org/10.1016/j.xphs.2022.12.022.
- Cioc, R. C., Joyce, C., Mayr, M., & Bream, R. N. (2023).Formation of N-Nitrosamine Drug Substance Related Impurities in Medicines: A Regulatory Perspective on Risk Factors and Mitigation Strategies.In Organic Process Research and Development.American Chemical Society. https://doi.org/10.1021/acs.oprd.3c00153.
- Wu, Y., Levons, J., Narang, A. S., Raghavan, K., & Rao, V. M. (2011).Reactive impurities in excipients: Profiling, identification and mitigation of drug-excipient incompatibility.In AAPS PharmSciTech (Vol.12, Issue 4, pp.1248-1263). https://doi.org/10.1208/s12249-011-9677-z.
- Berardi, A., Jaspers, M., & Dickhoff, B. H. J. (2023).Modeling the Impact of Excipients Selection on Nitrosamine Formation towards Risk Mitigation.Pharmaceutics, 15(8). https://doi.org/10.3390/pharmaceutics15082015.
- Boetzel, R., Schlingemann, J., Hickert, S., Korn, C., Kocks, G., Luck, B., Blom, G., Harrison, M., François, M., Allain, L., Wu, Y., & Bousraf, Y. (2023).A Nitrite Excipient Database: A Useful Tool to Support N-Nitrosamine Risk Assessments for Drug Products.Journal of Pharmaceutical Sciences, 112(6), 1615–1624. https://doi.org/10.1016/j.xphs.2022.04.016.
- Wang, Q. H., Yu, L. J., Liu, Y., Lin, L., Lu, R. gang, Zhu, J. ping, He, L., & Lu, Z. L. (2017).Methods for the detection and determination of nitrite and nitrate: A review.In Talanta (Vol.165, pp.709-720).https://doi.org/10.1016/j.talanta.2016.12.044.
- Jireš, J., & Douša, M. (2022).Nitrites as precursors of N-nitrosation in pharmaceutical samples – A trace level analysis.Journal of Pharmaceutical and Biomedical Analysis, 213.https://doi.org/10.1016/j.jpba.2022.114677.
720008197ZH,2024年2月