采用盐塞进样法改进大分子核酸的阴离子交换分析
摘要
随着核酸类药物的产品管线蓬勃发展,业界亟需经过改进的分析方法来快速确认各种大分子核酸的浓度、完整性和相对丰度。特别值得一提的是,阴离子交换法用于测定mRNA浓度和完整性的潜力相当可观,但目前的方法往往有严重的残留问题,导致应用该技术的可行性不高。本研究证明了谨慎选择色谱柱技术的重要性,还证明了Gen-Pak™ FAX弱阴离子交换色谱柱可以作为有效的起始色谱柱用于一种新的分析方法。此外,可通过以下三种方式来减少残留:缩短分析物滞留在色谱柱上的时间(1);以较高的配对离子浓度(高达100 mM)作为梯度起点(2);采用一种特殊的进样方式,用调节盐塞括住吸取的样品。通过这项研究,我们还弄清了单链mRNA变性与残留、选择性和分离度之间的关系。随着流动相温度升至自折叠的解链温度,选择性增加,但残留也随之增加。
优势
- 降低连续进样之间的残留
- 提高mRNA样品的回收率
- 改善重复性和方法稳健性
简介
近年来,生物制药行业对基因治疗药物的关注激增。合成核酸为开发新型疫苗和蛋白质表达调节药物开辟了一条新途径。最近,体外转录mRNA得到了广泛关注1,2。
完整mRNA原料药的分析需要使用液相色谱(LC)技术3。 一般使用离子对反相色谱(IP-RP)或体积排阻色谱(SEC)进行分析级分离,以检测异质性或检查分子完整性3。
尽管寡核苷酸和核酸的大规模纯化经常采用阴离子交换(AEX)分离技术,迄今为止有文献可查的分析级AEX分离方法却只有寥寥几种。AEX的分析级应用很有限,可能要归咎于方法稳健性不理想,例如使用此法分析大分子核酸时经常会观察到低回收率和高残留效应。Bridonneau及其同事发表的文章中提到,为了防止RNA适体纯化的AEX色谱运行之间发生残留,必须在专用条件下平衡系统30 min并运行空白样品4。 Guilherme等人提出了一些减少残留的解决方案,例如向洗脱缓冲液中添加螯合剂和/或变性剂(例如尿素、甲酰胺或异丙醇)来抑制分子间相互作用5。 还有研究人员提出使用高电荷态的阳离子分子(即亚精胺)作为添加剂,据信这种添加剂能稳定致密的超螺旋结构,从而提高回收率6。 最近有研究表明,采用弱离子对阳离子梯度的AEX方法得到了令人期待的mRNA分离效果7。 与传统的盐梯度相比,这种被称为“离子对阴离子交换”(IPAX)的方法回收率和选择性更高。除了溶质离子与固定相键合相之间的非特异性相互作用外,色谱柱硬件材料本身也可能导致低回收率和高残留。针对目前的情况,Minkner等人认为,AEX对siRNA的回收率能达到95%以上已属性能“优异”,据文献报道,不仅AEX,亲和色谱及疏水作用色谱(HIC)对大分子mRNA的回收率也不及小分子mRNA8,9。
考虑到上述因素,我们认为,如果能够成功改进AEX色谱分离方法,将有望为分析完整大分子核酸样品提供一种可靠的新方案。本文提出了一些有效的解决方案,能显著减少AEX分离中发生的残留。
实验
样品和流动相制备
ClenCap 5 moU EPO mRNA(长度:858个核苷酸)、荧光素酶(LUC) mRNA(长度:1929个核苷酸)和Cas9 mRNA(长度:4521个核苷酸)购自TriLink Biotechnologies(美国加利福尼亚州圣地亚哥)。用水将样品稀释至25 µg/mL后进样分析,无需进一步前处理。
三羟甲基氨基甲烷(TRIS)、盐酸胍(Gdn-HCl)和溴化钠(NaBr)购自Sigma-Aldrich(瑞士布克斯)。将Tris缓冲液制备为25 mM溶液,pH调节至约7.6,该25 mM Tris缓冲液用作流动相A。将2 M Gdn-HCl或2 M NaBr溶于25 mM Tris缓冲液中,制得流动相B。
执行盐塞进样时,可使用流动相B或溶于稀释10倍的BioResolve™ CX B浓缩液(P/N:186009064)的2 M NaBr溶液。
液相色谱条件
|
液相色谱系统: |
ACQUITY™ UPLC™ H-Class PLUS Bio系统(四元) |
|
检测: |
UV检测(波长260 nm) |
|
样品瓶: |
聚丙烯样品瓶(P/N:186002639) |
|
色谱柱: |
Gen-Pak FAX阴离子交换色谱柱, 2.5 µm, 4.6 mm x 100 mm(P/N:WAT015490) |
|
柱温: |
环境温度到 45 ℃ |
|
样品温度: |
5 ℃ |
|
进样体积: |
2.0 µL(样品) |
|
括号式进样顺序: |
1.0 μL(前盐塞)+ 2.0 μL(样品)+ 1.0 μL(后盐塞) |
|
流速: |
0.6 mL/min |
|
流动相A: |
25 mM TRIS水溶液(pH = 7.6) |
|
流动相B: |
2 M盐酸胍(Gdn-HCl)的25 mM TRIS溶液(pH = 7.6)或 2 M溴化钠(NaBr)的25 mM TRIS溶液(pH = 7.6) |
|
梯度: |
推荐用于快速分离的陡峭梯度: - 如果使用2 M Gdn-HCl流动相:流动相B在6 min内从0增加到25% - 如果使用2 M NaBr流动相:流动相B在7 min内从15%增加到50% 推荐用于提高选择性的缓梯度: - 如果使用2 M NaBr流动相:流动相B在15 min内从12%增加到35% |
色谱柱钝化
使用至少20~50倍柱体积的流动相平衡色谱柱。然后使用目标样品连续执行几次(3~4次)高载样量(例如5~10 μg)进样,钝化固定相的活性位点。
结果与讨论
分析生物分子(大分子)时的进样重复性差、残留效应高,背后的原因通常是这类分子与表面之间发生了不良次级相互作用,以及大分子系统内部发生了分子间和分子内相互作用。大分子通常具有表面活性,它们与不同类型的表面接触时,会发生非特异性吸附10。 这是一个至关重要的过程,因为它可能导致溶质损失或发生聚集。在吸附事件中,大多数大分子的形状都会发生变化(构象变化,例如展开)。吸附剂表面被大分子占据的区域通常称为“足迹”(the footprint)11。 足迹通常随滞留时间增加而增加,此过程可称为“展开过程”。足迹导致的额外吸附通常是部分可逆的12,13。 吸附动力学和足迹表面积在很大程度上还取决于溶质浓度14。 分析物浓度较高时,样品表面会在短时间内被占据,因此可用于展开的时间也会更短。这样一来,平均足迹就比较小,但吸附浓度较高。溶剂pH和离子强度等其他参数也可能影响足迹大小和展开过程。
已经有多项研究报道了上面提到的一些影响15-18。
滞留时间和初始流动相强度的影响
初步实验表明,mRNA与固定相结合的时长可能与残留有关。
已有研究证实,在AEX分离中,mRNA遵循类似“开-关”式(结合-洗脱)的洗脱机制7。 因此,我们可以合理地假设mRNA会结合在色谱柱柱头并滞留于此,直到它们经历流动相组成的梯度洗脱。利用这段时间,分析物可以展开并与固定相发生多点相互作用。人们可能会觉得滞留时间越短,结合区段就会越少,这样一来,吸附相互作用就越弱,残留也就越低。
我们通过实验系统地研究了滞留时间对残留的影响。设定流动相B在6 min内从0增加到25%的Gdn-HCl梯度,并在梯度开始时设定不同的初始等度保持时间(0% B)。也就是说,将初始等度保持时间设置为0、0.5、1、2、4和8 min。进样EPO和Cas9 mRNA样品,并测定样品进样之后的空白进样的残留(以百分比计)。图1A显示了残留随溶质结合时间的变化。该图表明残留与结合时间之间确实存在相关性。结合时间越短,残留越低。和预期一致,这些实验表明应通过缩短分析运行时间来降低残留。将滞留时间限制在3 min左右时,残留低至4%~8%,相比之下,长梯度条件下的残留高达10%~20%。
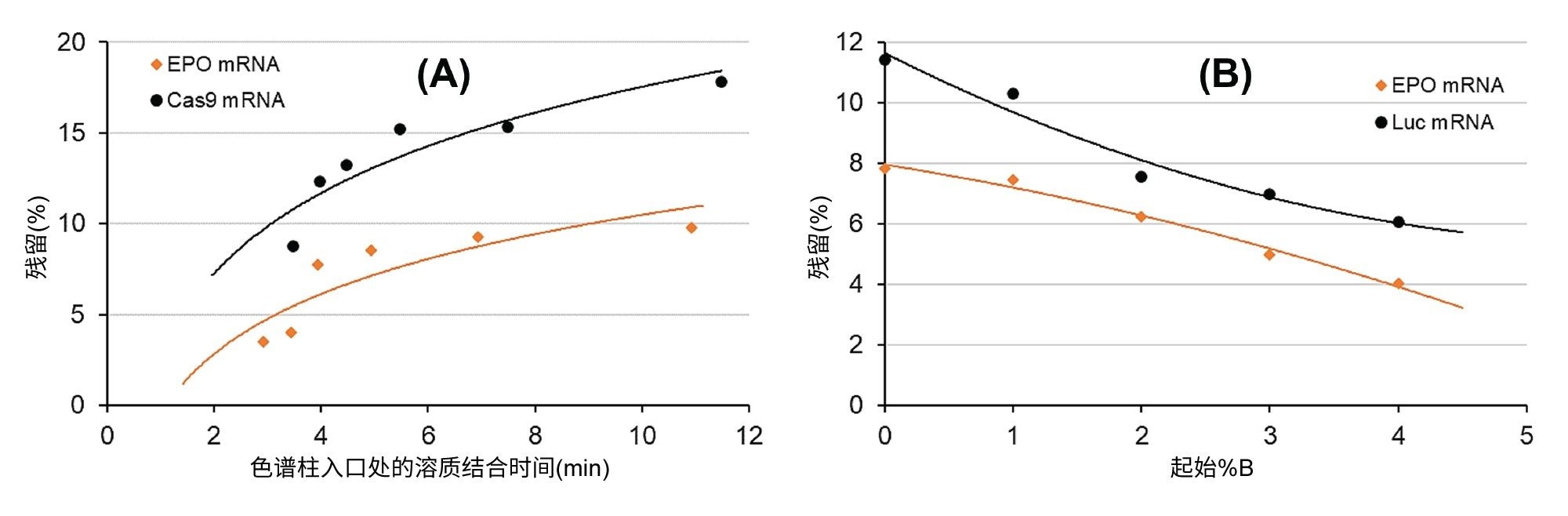 图1.mRNA溶质结合(滞留)时间(A)和初始流动相强度(B)对AEX残留的影响。色谱柱:Gen-Pak FAX 100 x 4.6 mm, 2.5 µm色谱柱,流动相A:25 mM TRIS,pH = 7.6,流动相B:25 mM TRIS,pH = 7.6 + 2 M Gdn-HCl,流速 = 0.6 mL/min,梯度:流动相B在6 min内从0增加到25%,环境温度(约22 ℃)。
图1.mRNA溶质结合(滞留)时间(A)和初始流动相强度(B)对AEX残留的影响。色谱柱:Gen-Pak FAX 100 x 4.6 mm, 2.5 µm色谱柱,流动相A:25 mM TRIS,pH = 7.6,流动相B:25 mM TRIS,pH = 7.6 + 2 M Gdn-HCl,流速 = 0.6 mL/min,梯度:流动相B在6 min内从0增加到25%,环境温度(约22 ℃)。
在另一个实验中,我们研究了流动相初始强度的影响。如果假设溶质展开发生在固定相表面上,那么有人可能会认为只要削弱初始结合时的相互作用,溶质展开就不会太严重(或展开较慢)。因此,本研究采用固定的梯度时间,并将流动相B的初始比例调整为0%、1%、2%、3%和4%。图1B显示了我们观察到的EPO和Luc mRNA残留随初始流动相组成的变化。由图可见明显的趋势,初始%B越高,残留就越低。从4% B(配对离子约80 mM)而不是0% B开始梯度可使残留减半。这一观察结果表明,应适当提高梯度开始时的流动相B比例(即50~100 mM配对离子),而不是从0%流动相B开始。
请注意,结合时间和起始%B的影响都取决于样品,因此需要针对目标样品进行优化。
用盐塞括住样品溶液段(括号式进样)
溶剂强度不匹配是一个术语,用于描述进样溶剂和流动相具有不同洗脱液强度的情况19。 当样品溶剂强于流动相组成时,溶剂强度不匹配的问题尤其突出。这种情况通常会导致部分或全部流穿效应,至少都会导致前沿峰或分裂峰19。 多维分离和亲水作用色谱(HILIC)分析经常会遇到这类问题。为了减弱强溶剂效应,可以应用专门编写的进样序列将样品引入色谱柱,用一前一后两个稀释过的样品塞“括”住吸取的样品。此类进样序列已被应用于改善分离性能或限制流穿效应20,21。
在mRNA的AEX分离中,结合相互作用本身就非常强,因此需要反转上述进样序列方案(通常用于提高初始结合相互作用的强度)。在这种情况下,样品应随强溶剂塞一起进样,以限制初始结合的强度。因此,我们建议用含有高浓度盐(配对离子)的溶剂塞括住样品。除了提高离子强度外,还可以将溶剂塞的pH调节至接近固定相官能团的pKa(如果使用弱阴离子交换剂,即调节至pH 10~11)。同样,这种组合效应是为了限制初始吸附的强度。
我们通过系统性实验确定了对进样序列而言最重要的因素,以及它们如何影响AEX分离中的mRNA残留。本研究考察了以下因素:(1)前盐塞体积;(2)后盐塞体积;(3)括号式盐塞的总体积;(4)所用盐的类型,包括NaCl、(NH4)2SO4、Gdn-HCl和NaBr,以及(5)盐塞的pH。
结果表明,最有利的进样顺序是用2 M NaBr溶液(pH约为10)形成的一前一后两个盐塞括住样品溶液段。图2显示了括号式进样的示意图。请注意,pH高于10.5~11.0时,mRNA可能会变性,碱基配对和碱基堆积相互作用可能会被破坏。因此,完整RNA在高pH条件下可能线性化,进而导致展开、结合足迹扩大以及显著更强的吸附。此外,某些核酸碱基在pH超过10的条件下还可能去质子化,给核酸分析物额外增加负电荷22。 这可能是高pH条件的AEX分离中mRNA保留性增加的原因所在3。 因此,pH过高(>11)的溶剂不适合用作盐塞,以往的研究曾观察到残留增加。
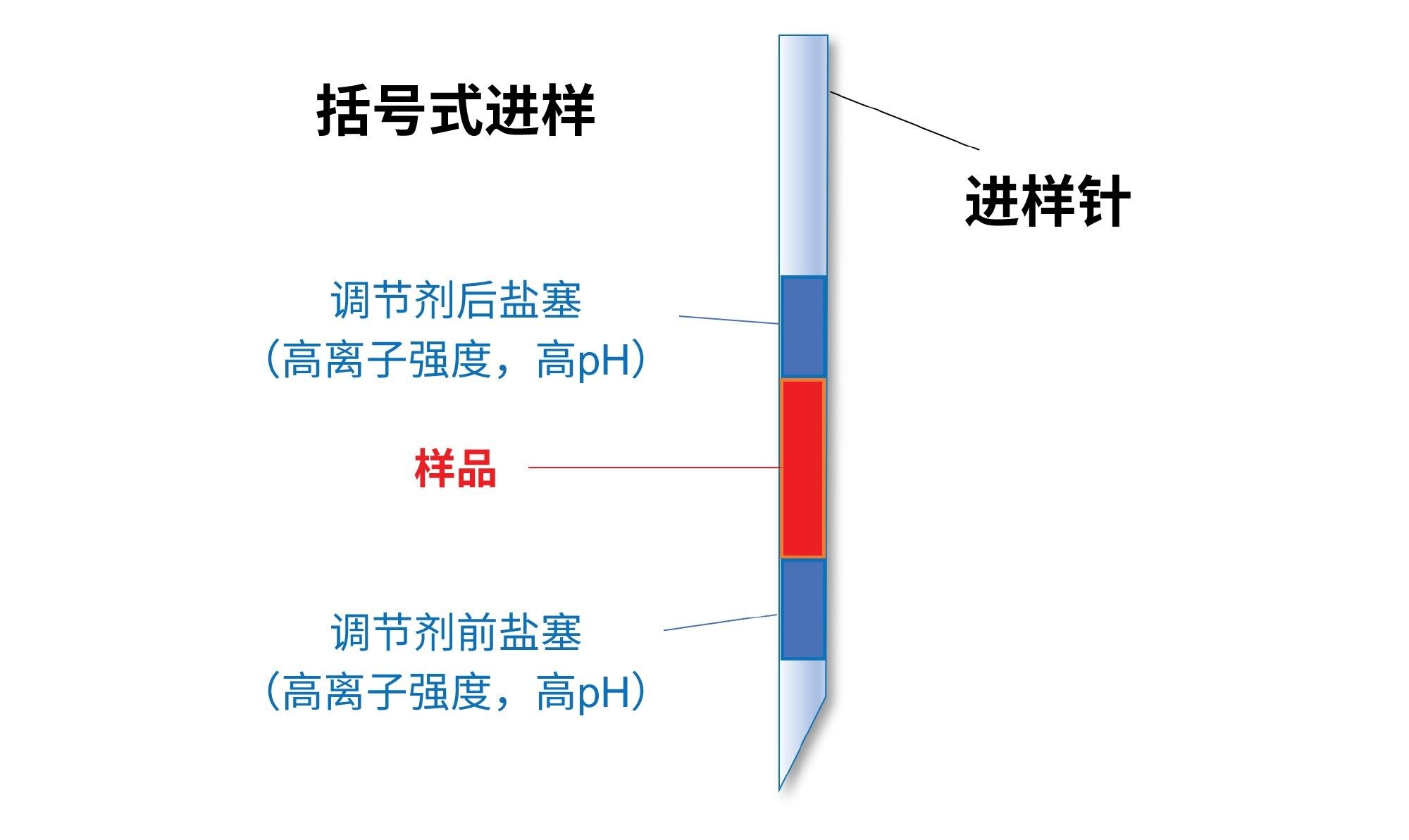 图2.进样序列示意图,在该图中,吸取的样品被高离子强度且高pH的调节剂盐塞括了起来。用户可以使用Empower™软件的“自动添加”选项为样品组创建他们想要的任何进样序列23。
图2.进样序列示意图,在该图中,吸取的样品被高离子强度且高pH的调节剂盐塞括了起来。用户可以使用Empower™软件的“自动添加”选项为样品组创建他们想要的任何进样序列23。
为了在避免样品流穿的同时尽可能降低残留,我们需要优化调节盐塞与样品溶液段的体积比。图3显示了EPO mRNA的残留和回收率如何随调节盐塞与样品的体积比(V调节剂/V样品)变化,适用于前盐塞和后盐塞体积相同的情况。由图可见,在(V调节剂/V样品)达到约1.2~1.3之前,残留减少且回收率增加。超出这个“限值”后,强调节盐塞会侵占一小部分进样的样品体积,导致色谱图上出现部分流穿峰。如果(V调节剂/V样品) ≥ 2,则整个mRNA峰都会在色谱柱的死时间处洗脱(完全流穿)。如果将(V调节剂/V样品)设置为大约1~1.2,残留可降至2%~3%,相比之下,不使用盐塞调节时通常会观察到10%~20%的残留。请注意,理想的(V调节剂/V样品)比可能取决于样品、进样器设备、系统体积、流动相和色谱柱,需要针对每种方法单独优化。将(V调节剂/V样品)设为1左右似乎是一个很好的起点。例如,如果要进样2 μL mRNA样品,使用1 μL调节剂前盐塞+ 2 μL样品+ 1 μL调节剂后盐塞编写成进样序列会是个不错的起点。
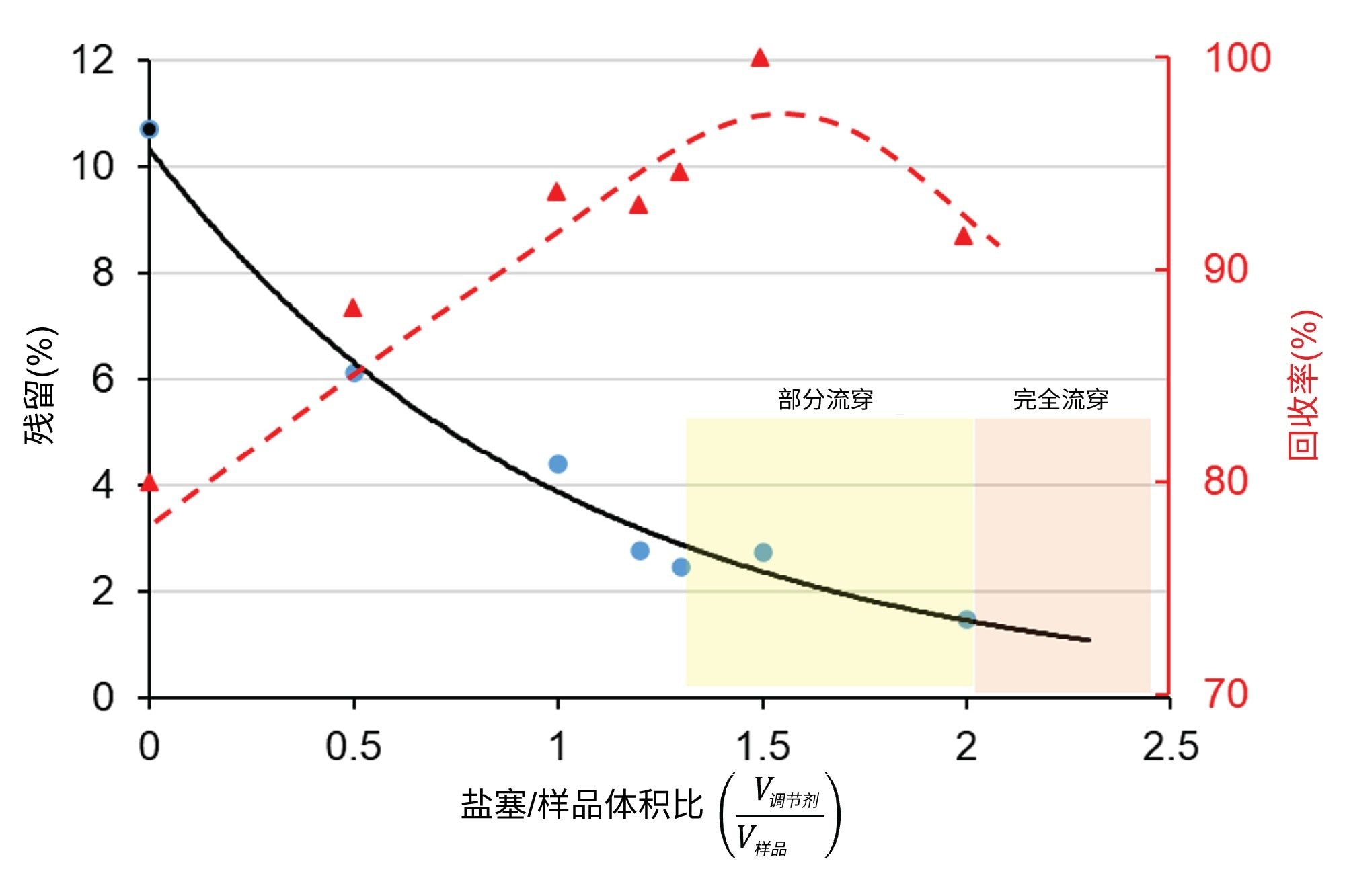 图3.调节剂盐塞与样品的体积比对残留(黑色曲线)和回收率(红色曲线)的影响。色谱柱:Gen-Pak FAX 100 x 4.6 mm, 2.5 µm色谱柱,流动相A:25 mM TRIS,pH = 7.6,流动相B:25 mM TRIS,pH = 7.6 + 2 M NaBr,流速 = 0.6 mL/min,梯度:流动相B在7 min内从15%增加到50%,环境温度(约22 ℃)。样品:EPO mRNA(进样2 μL),调节剂盐塞:流动相B。
图3.调节剂盐塞与样品的体积比对残留(黑色曲线)和回收率(红色曲线)的影响。色谱柱:Gen-Pak FAX 100 x 4.6 mm, 2.5 µm色谱柱,流动相A:25 mM TRIS,pH = 7.6,流动相B:25 mM TRIS,pH = 7.6 + 2 M NaBr,流速 = 0.6 mL/min,梯度:流动相B在7 min内从15%增加到50%,环境温度(约22 ℃)。样品:EPO mRNA(进样2 μL),调节剂盐塞:流动相B。
与色谱柱(固定相)相关的残留
我们研究了包括弱AEX固定相和强AEX固定相在内的多种固定相,只发现细微差别,似乎弱交换剂的残留通常略低。但需要注意的是,残留始终取决于样品和分析条件。即使在不同的弱离子交换剂之间也可能观察到差异,这可能与键合相密度、键合相可及性(形态)差异,以及可能存在的其他相互作用(如氢键)有关。
图4比较了Gen-Pak FAX弱阴离子交换色谱柱和某款弱阴离子交换整体柱,这两根色谱柱均采用经过优化的同一种括号式进样方法运行分析。Gen-Pak FAX色谱柱的残留明显更低,分离度与另一根色谱柱相当,某些峰的锐度还得到了显著改善。
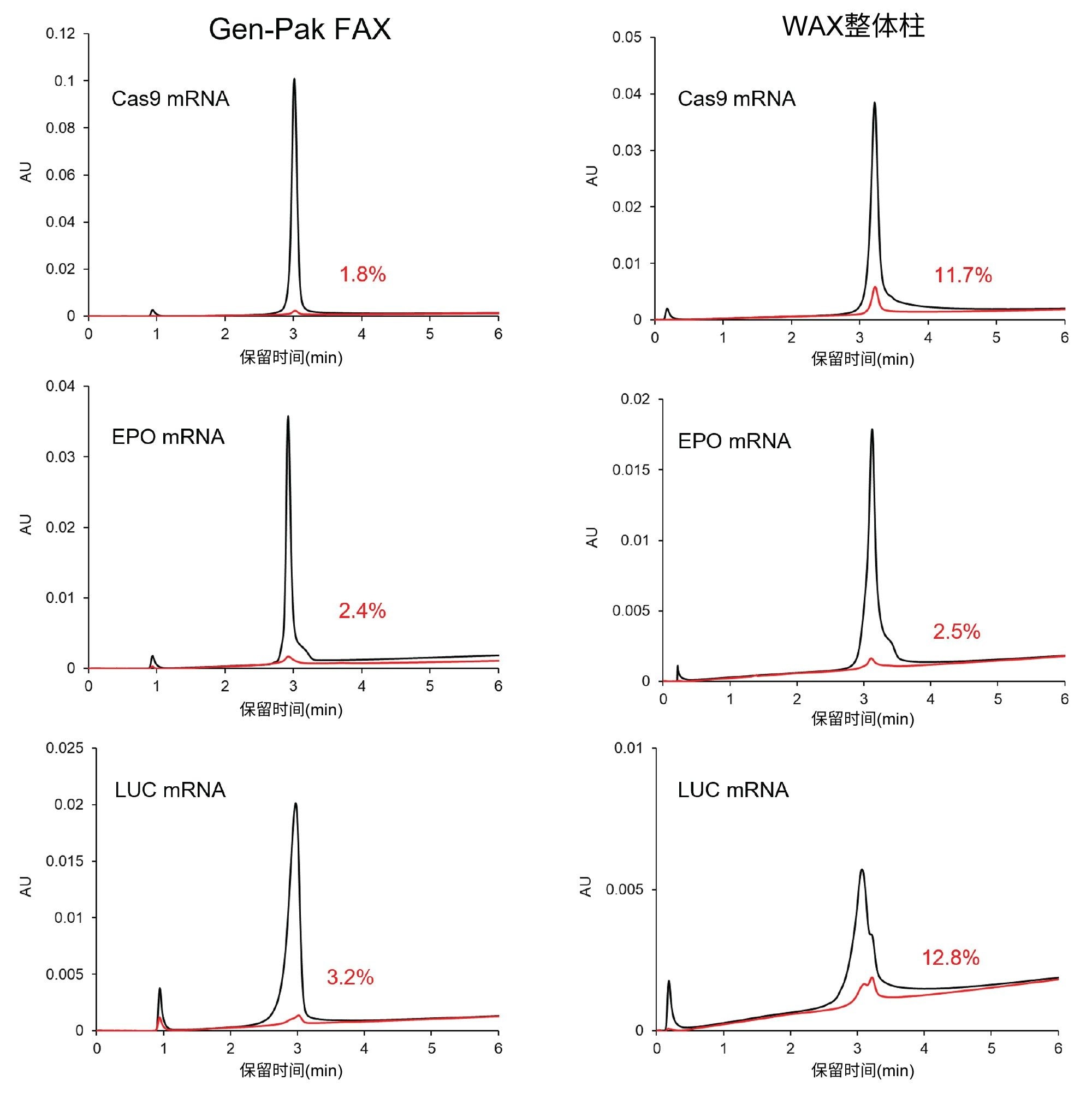 图4.两根色谱柱上观察到的残留对比。色谱柱:Gen-Pak FAX 100 x 4.6 mm, 2.5 µm色谱柱(左图)和WAX 4.95 x 5.2 mm整体柱(右图)。梯度条件:与图3相同,调节剂溶剂塞:2 M NaBr溶于pH 10.2的缓冲液。括号式进样:1 μL调节剂前盐塞+ 2 μL样品+ 1 μL调节剂后盐塞。以红色显示的百分比数值表示在样品进样之后执行的空白进样中观察到的残留百分比。
图4.两根色谱柱上观察到的残留对比。色谱柱:Gen-Pak FAX 100 x 4.6 mm, 2.5 µm色谱柱(左图)和WAX 4.95 x 5.2 mm整体柱(右图)。梯度条件:与图3相同,调节剂溶剂塞:2 M NaBr溶于pH 10.2的缓冲液。括号式进样:1 μL调节剂前盐塞+ 2 μL样品+ 1 μL调节剂后盐塞。以红色显示的百分比数值表示在样品进样之后执行的空白进样中观察到的残留百分比。
温度影响
已有研究表明,在AEX色谱分离中,升高温度会导致RNA样品中检出的峰变少,但峰形更清晰24。 这归因于二级结构减少。此外,在较高温度下还观察到保留性增加,这也可以解释为内部结构被破坏的结果。最近,我们发现温度对AEX中的mRNA回收率和残留也有重大影响7。 温度效应导致的残留变化难以预测,因为许多参数都可能改变mRNA、水相流动相和固定相之间的相互作用的强度。在存在大量盐的情况下,疏溶剂效应、盐析、盐溶、mRNA脱水和结构重排全都可能发生,只是程度各不相同7。 因此,我们很有兴趣研究温度对括号式进样的影响。图5显示了在环境温度和高温下(温度 = 35 ℃和45 ℃)进样EPO和Cas9 mRNA样品得到的色谱图。残留似乎随温度升高而增加,这与我们观察到的保留性增强趋势一致(表明高温下的结合更强)。因此,考虑到样品残留,优选在环境温度下执行实验。但是,峰形、选择性和分离特征会随温度发生显著变化,并且高温似乎还对样品组分的分离度产生了一些有利影响。最终解决方案是考虑设置两种方法,一种在室温条件下运行,另一种在较高的柱温下运行。室温(低温)方法可能适用于测定含量/浓度,而高温方法对于研究mRNA的生物物理特性和化学异质性可能有价值。
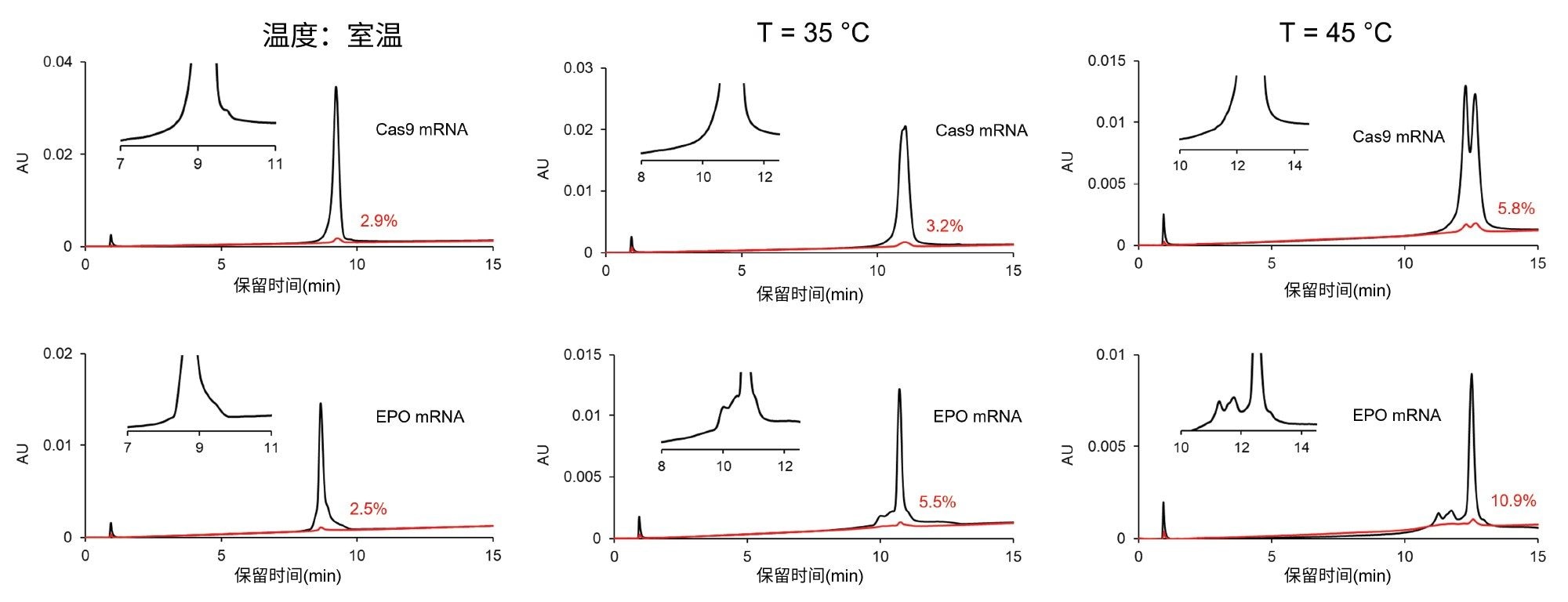 图5.温度对色谱特征(选择性)和残留的影响。色谱柱:Gen-Pak FAX 100 x 4.6 mm, 2.5 µm色谱柱,流动相A:25 mM TRIS,pH= 7.6,流动相B:25 mM TRIS,pH = 7.6 + 2 M NaBr,流速 = 0.6 mL/min,梯度:流动相B在15 min内从12%增加到35%(缓梯度),调节剂溶剂塞:流动相B。括号式进样:1 μL调节剂前盐塞+ 2 μL样品+ 1 μL调节剂后盐塞。温度:室温(左)、35 ℃(中)和45 ℃(右)。
图5.温度对色谱特征(选择性)和残留的影响。色谱柱:Gen-Pak FAX 100 x 4.6 mm, 2.5 µm色谱柱,流动相A:25 mM TRIS,pH= 7.6,流动相B:25 mM TRIS,pH = 7.6 + 2 M NaBr,流速 = 0.6 mL/min,梯度:流动相B在15 min内从12%增加到35%(缓梯度),调节剂溶剂塞:流动相B。括号式进样:1 μL调节剂前盐塞+ 2 μL样品+ 1 μL调节剂后盐塞。温度:室温(左)、35 ℃(中)和45 ℃(右)。
结论
已知大分子核酸的阴离子交换分析可能会出现高残留和低回收率的问题。本文提出了一些新的方法考量因素,希望能减少残留,从而提高方法稳健性。过去的多轮研究已经确认,Gen-Pak FAX色谱柱分离某些大分子单链核酸样品的效果相当出众。这为实施新方法提供了可靠的依据。
研究发现,就残留而言,缩短分析时间可以起到改善作用。以相对较高的洗脱液强度作为梯度起点也很有帮助。
不过,最有用的方法当属应用专门编写的样品进样序列,用我们所谓的“调节剂塞”括住样品。这些溶剂塞含有高浓度的盐,并用缓冲液将pH调节为10左右。这种括号式进样有助于降低色谱柱柱头处的溶质结合强度,从而提高回收率和降低残留。调节剂盐塞的体积和比率需要针对每种方法单独优化。本研究证明,将(V调节剂/V样品)设为1左右的效果很好。通过结合这种创新的括号式进样模式与Gen-Pak FAX色谱柱,我们成功将大分子mRNA的残留降至约2%,相比之下,传统AEX方法的残留高达10%~20%。
参考资料
- C. Rinoldi, S.S. Zargarian, P. Nakielski, X. Li, A. Liguori, F. Petronella, D. Presutti, Q. Wang, M. Costantini, L. De Sio, C. Gualandi, B. Ding, F. Pierini.Nanotechnology-assisted RNA delivery: from nucleic acid therapeutics to COVID-19 vaccines. Small Methods 5 (2021) 2100402, https://doi.org/10.1002/smtd.202100402
- A. Akinc, M.A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M.J. Hope, T.D. Madden, B.L. Mui, S.C. Semple, Y.K. Tam, M. Ciufolini, D. Witzigmann, J.A. Kulkarni, R. van der Meel, P.R. Cullis.The Onpattro story and the clinical translation of nanomedicines containing nucleic acid based drugs. Nat. Nanotechnol.2019, 14, 1084–1087.https://doi.org/10.1038/s41565-019-0591-y
- S. Fekete, C. Doneanu, B. Addepalli, M. Gaye, J. Nguyen, B. Alden, R. Birdsall, D. Han, G. Isaac, M. Lauber.Challenges and emerging trends in liquid chromatography-based analyses of mRNA pharmaceuticals.J. Pharm.Biomed.Alna., 224 (2023) 115174. https://doi.org/10.1016/j.jpba.2022.115174
- P. Bridonneaua, S. Bunch, R. Tengler, K. Hill, J. Carter, W. Pieken, D. Tinnermeier, R. Lehrman, D.W. Drolet.Purification of a highly modified RNA-aptamer: Effect of complete denaturation during chromatography on product recovery and specific activity. J. Chromatogr. B, 726 (1999) 237–247.
- G N Ferreira, G A Monteiro, D M Prazeres, J M Cabral.Downstream processing of plasmid DNA for gene therapy and DNA vaccine applications. Trends Biotechnol. 18 (2000) 380–388.https://doi.org/10.1016/s0167-7799(00)01475-x
- J.C. Murphy, J.A. Wibbenmeyer, G.E. Fox, R.C. Willson.Purification of plasmid DNA using selective precipitation by compactation agents. Nat. Biotechnol. 17 (1999) 822–823.https://doi.org/10.1038/11777
- S. Fekete, H. Yang, K. Wyndham, M. Lauber.Salt gradient and ion-pair mediated anion exchange of intact messenger ribonucleic acids. J. Chromatogr. Open, 2 (2022) 100031.https://doi.org/10.1016/j.jcoa.2022.100031
- R. Minkner, J. Boonyakida, E.Y. Park, H. Wätzig.Oligonucleotide separation techniques for purification and analysis: What can we learn for today’s tasks?. Electrophoresis, 43 (2022) 2402–2427.https://doi.org/10.1002/elps.202200079
- T. Cui, K. Fakhfakh, H. Turney, G. Güler-Gane, A. Toloczko, M. Hulley, R. Turner.Comprehensive studies on building a scalable downstream process for mRNAs to enable mRNA therapeutics. Biotechnol. Prog.39 (2023) e3301.https://doi.org/10.1002/btpr.3301
- K. Höger, T. Becherer, W. Qiang, R. Haag, W. Friess, S. Küchler.Polyglycerol coatings of glass vials for protein resistance.Eur.J. Pharm.Biopharm. 85(2013) 756–764.https://doi.org/10.1016/j.ejpb.2013.04.005
- M Rodriguez-Allera, A. Cusumano, A. Beck, D. Guillarme, S. Fekete.Importance of vial shape and type on the reproducibility of size exclusion chromatography measurement of monoclonal antibodies. J. Chromatogr. B, 1032 (2016) 131–138.http://dx.doi.org/10.1016/j.jchromb.2016.04.032
- J. Mathes, Protein Adsorption to Vial Surfaces—Quantification, Structural andMechanistic Studies, Ph.D. thesis, Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität, München, 2010.
- J.M. Kleijn, W. Norde.The adsorption of proteins from aqueous solution on solid surfaces. Het. Chem.Rev. 2 (1995) 157–172.
- M.V.D. Veen, M.C. Stuart, W. Norde, Spreading of proteins and its effect on adsorption and desorption kinetics. Colloids Surf. B 54 (2007) 136–142.https://doi.org/10.1016/j.colsurfb.2006.08.017
- S. Fekete, A. Murisier, D. Guillarme.Hydrophobic Interaction Chromatography (HIC) for the Characterization of Therapeutic Monoclonal Antibodies and Related Products, Part 1: Theoretical Aspects. LCGC Europe, 34 [2021] 101–105.
- E. Haimer, A. Tscheliessnig, R. Hahn, A. Jungbauer.Hydrophobic interaction chromatography of proteins IV: Kinetics of protein spreading. J. Chromatogr. A.1139 (2007) 84–94.https://doi.org/10.1016/j.chroma.2006.11.003
- R. Ueberbacher, E. Haimer, R. Hahn, A. Jungbauer.Hydrophobic interaction chromatography of proteins V. Quantitative assessment of conformational changes. J. Chromatogr. A.1198–1199 (2008) 154–163.https://doi.org/10.1016/j.chroma.2008.05.062
- J.A. Queiroz, C.T. Tomaz, J.M.S. Cabral.Hydrophobic interaction chromatography of proteins. J. Biotechnol. 87 (2001) 143–159.https://doi.org/10.1016/S0168-1656(01)00237-1
- S. Chapel, S. Heinisch.Strategies to circumvent the solvent strength mismatch problem in online comprehensive two-dimensional liquid chromatography. J. Sep. Sci., 45 (2022) 7–26. https://doi.org/10.1002/jssc.202100534
- A. C. Sanchez, J.A. Anspach, T. Farkas.Performance optimizing injection sequence for minimizing injection band broadening contributions in high efficiency liquid chromatographic separations. J. Chromatogr. A, 1228 (2012) 338–348.https://doi.org/10.1016/j.chroma.2012.01.038
- M. Taylor, J. Kawakami, D.V. McCalley.Managing sample introduction problems in hydrophilic interaction liquid chromatography. J. Chromatogr. A, 1700 (2023) 464006.https://doi.org/10.1016/j.chroma.2023.464006
- S. Ganguly, K.K. Kundu.Protonation/deprotonation energetics of uracil, thymine, and cytosine in water from e.m.f./spectrophotometric measurements.Can. J. Chem.72 (1994) 1120–1126.
- https://support.waters.com/KB_Inf/Empower_Tips_of_the_Week/WKB121702_In-Needle_Auto_Additions_in_Empower
- A. Kanavarioti.HPLC methods for purity evaluation of man-made single-stranded RNAs. Sci Rep 9 (2019) 1–13. https://doi.org10.1038/s41598-018-37642-z.
720007991ZH,2023年7月