离子对反相液相色谱法分析mRNA Poly(A)尾异质性
摘要
优势
- 稳定的poly(A)尾异质性分析方法
- 适用于质量控制检测的LC UV方法
- 能够使长度多达150个寡核苷酸的poly(A)尾异构体获得n/n-1分离度
简介
治疗性信使mRNA分子为单链核酸,长度通常为2,000–10,000个核苷酸。治疗性mRNA的结构由若干个不同的部分组成,包括修饰的5'端帽、3'和5'非翻译区(UTR)、mRNA编码区或基因转录序列以及在3'端的poly(A)尾(见图1)。5'端帽可大幅减少RNA降解,同时通过固定细胞中的核糖体而促进翻译过程。3'和5'非翻译区直接影响翻译,通常优化此区域的序列以实现尽可能高的RNA翻译效率。mRNA编码区编码目的基因(GOI)。此区域被翻译成所需蛋白质,且通常包含修饰的核苷酸(通常为N1-甲基假尿苷),以降低清除率并提高蛋白质产量。最后,poly(A)尾存在于3'端,此区域在大幅减少RNA降解和维持mRNA的体内稳定性方面起关键作用。mRNA疫苗中poly(A)尾的长度通常为100–150个核苷酸(nt)。
SARS-CoV-2疫苗的成功加快了有关mRNA疗法的研究活动。目前有多项mRNA临床试验正在进行中。mRNA疗法的研究、开发和生产需要稳定的分析方法。离子对反相液相色谱(IP RP LC)法是寡核苷酸分析的理想选择[1–5]。 该方法可使长度多达约60 nt的物质获得n/n-1分离度,但是随着寡核苷酸和mRNA的长度增加,难度逐渐攀升。即便使用先进的分离方法和质谱(MS)技术,分析mRNA的poly(A)尾异质性也是一项艰巨的任务。完整mRNA分子在分析之前通常先用选择性核酸酶(例如核糖核酸酶T1)酶解。核糖核酸酶T1在G核苷酸的3'端裂解RNA序列[6],产生相对较短的寡核苷酸混合物,随后可通过LC MS/MS进行分析[1,5,7]。mRNA的poly(A)尾部不包含将会裂解的G核苷酸。因此,3’ poly(A)尾以长度为100–150 nt的寡核苷酸形式从mRNA中释放出来(见图1)。分离长度相差单个核苷酸的长链寡核苷酸对聚丙烯酰胺凝胶电泳[8]、毛细管凝胶电泳[9,10]或色谱方法均构成挑战[8,9,10,11,13]。
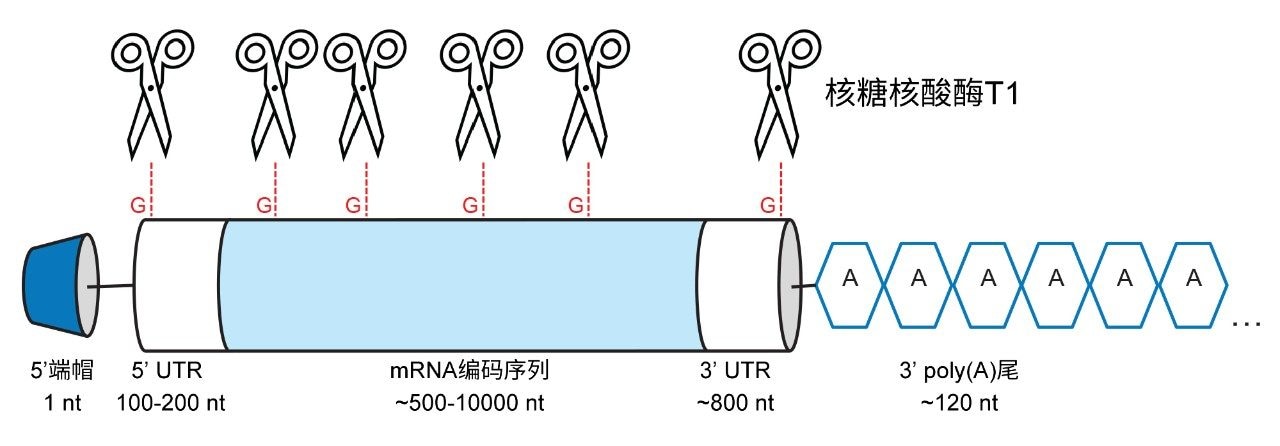 图1.mRNA结构示意图。剪刀指示核糖核酸酶T1的推断性裂解位点。酶解产生短链RNA寡核苷酸并释放出3'端poly(A)尾。
图1.mRNA结构示意图。剪刀指示核糖核酸酶T1的推断性裂解位点。酶解产生短链RNA寡核苷酸并释放出3'端poly(A)尾。
本应用纪要介绍了一种分析poly(A)尾异质性的高分离度IP RP LC方法。用核糖核酸酶T1释放的poly(A)尾通过IP RP LC分离为寡核糖核苷酸腺苷。该IP RP LC方法使长达约150 nt的寡核苷酸获得了n/n-1分离度。该方法可用于分析poly(A)尾异质性并鉴定poly(A)尾中主要寡核苷酸的长度。或者,也可使用简单的体积排阻色谱法搭配紫外检测(SEC UV)来估计poly(A)尾的平均长度。SEC UV方法另在一份应用纪要720007853中说明。
实验
样品前处理
EPO mRNA标准品购自TriLink Biotechnologies,部件号L-7209。EPO mRNA包含Cap 1 5'结构,其经过聚腺苷酸化,并且整个序列中包含修饰的5-甲氧基尿苷。Fluc-beta mRNA样品购自AmpTec,部件号M1436/1000-C1-A120-NM-P0。此mRNA经过聚腺苷酸化,并且序列中包含未修饰的尿苷。按照以下方案,用核糖核酸酶T1(Thermo Fisher,部件号EN0542,1000 U/µL)对1 mg/mL mRNA样品溶液进行酶解:将50 µL 1 mg/mL mRNA与10 µL rCutSmart 10x缓冲液(New England Biolabs,部件号B6004S)混合,并加入2 µL核糖核酸酶T1。将样品在37 °C下酶解30 min。酶解后,加入1 µL Quick CIP酶(New England Biolabs,部件号M0525S,5000 µ/mL),以去除寡核苷酸的3'磷酸基团。去磷酸化在室温下进行30 min。向样品中加入20 µL 1M TEAA(Sigma,部件号90358)和137 µL不含核糖核酸酶的水(Sigma,部件号W4502)。按照以下方案,使用Oasis™ HLB 96孔µElution板(Waters™,部件号186001828BA)净化最终体积约200 µL的样品:依次用50 µL乙腈和100 µL 0.1 M TEAA溶液清洗孔板。使用约0.5 mL/min的流速上样。收集样品并二次上样。用40 μL 30%乙腈溶液将样品从孔板上洗脱下来。取1–5 µL洗脱液进样至离子对反相液相色谱柱上进行分析。100 nt寡核糖核苷酸腺苷作为定制寡核苷酸购自IDT DNA(美国艾奥瓦州科尔维尔)。
IP RP LC条件
|
液相色谱系统: |
ACQUITY™ Premier UPLC™(配备SM-FTN和QSM) |
|
检测: |
PDA,Ti 5 µL流通池,260 nm |
|
色谱柱: |
ACQUITY Premier BEH C18寡聚核苷酸分析专用柱, 300 Å, 2.1 × 150 mm, 1.7 µm(P/N:186010541) |
|
柱温: |
60 °C |
|
样品温度: |
10 °C |
|
进样体积: |
1.0 µL(样品) |
|
流速: |
0.3 mL/min |
|
流动相A: |
100 mM乙酸辛铵(OAA)的40%乙腈和1%六氟异丙醇(HFIP)溶液,v:v |
|
流动相B: |
100 mM OAA的90%乙腈和1% HFIP溶液 |
|
流动相D: |
100%乙腈 |
|
梯度: |
40 min内46% A、54% B和0% D分别变为28% A、62% B和10% D |
|
色谱软件: |
Empower™ v 3.0 |
流动相A的制备:将116.13 g水与60.86 g乙腈混合;加入1.145 mL冰醋酸、3.305 mL辛胺和2 mL HFIP。充分混合。
流动相B的制备:将19.355 g水与136.92 g乙腈混合;加入1.145 mL冰醋酸、3.305 mL辛胺和2 mL HFIP。充分混合。
结果与讨论
IP RP LC方法开发策略
寡核苷酸的IP RP LC分离利用了离子对流动相添加剂[1,2,14]。 IP添加剂(例如烷基胺)会吸附到疏水性RP吸附剂上,促进带正电荷的烷基胺与寡核苷酸上带负电荷的磷酸基团发生离子相互作用。IP RP LC虽然能够轻松分离15–30 nt的短链寡核苷酸,但要分离长度40–60 nt的寡核苷酸已经较为困难,分离60–100 nt的寡核苷酸则更加困难[13]。 这是因为10 nt与11 nt寡核苷酸的电荷差异有10%(分别含有9个或10个磷酸基团),而100 nt和101 nt寡核苷酸的电荷差异仅1%,因此100/101 nt的分离选择性降低。探索长链寡核苷酸分离度优化的策略如下。(i)使用高效疏水性烷基胺(己胺、辛胺)[1.14]。 (ii)在流动相中加入高浓度的IP试剂(例如50–100 mM)。(iii)使用高效色谱柱(填充亚2 μ颗粒的长色谱柱)。(iv)使用缓梯度,例如每分钟0.1–0.5%乙腈[2]。 我们将ACQUITY Premier BEH C18寡核苷酸分析专用柱(300 Å, 2.1 × 150 mm, 1.7 µm)与100 mM乙酸辛铵IP流动相和每分钟0.25%乙腈的梯度结合使用,使长达约150 nt的poly(A)尾寡核苷酸获得了理想的分离度。不建议OAA浓度高于100 mM,因为洗脱时使用的乙腈浓度高于80%,可能会导致核酸发生柱上沉淀。市面上有些mRNA样品含有对IP RP LC色谱柱性能不利的成分,因此,在进行IP RP LC分析之前,建议先用Oasis HLB 96孔µElution板净化样品。
IP RP LC方法在poly(A)尾分析中的应用
图2展示了经优化用于核糖核酸酶T1酶解后EPO mRNA (TriLink)和Fluc Beta mRNA (AmpTech)分析的IP RP LC方法。酶解产生的2–30 nt短链寡核苷酸在五分钟前洗脱(数据未显示)。图2突出显示了在12~35 min之间洗脱的poly(A)物质。长度为124 nt的峰组(蓝色色谱图)代表EPO mRNA poly(A)尾的分析结果。红色色谱图显示Fluc Beta mRNA poly(A)尾稍长,集中在128 nt峰附近。
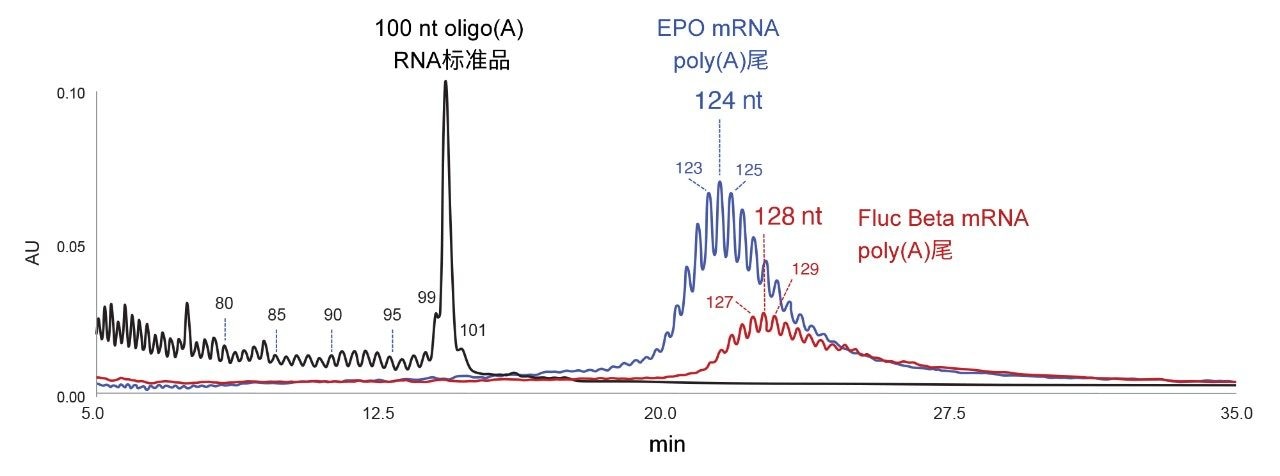 图2.IP RP LC方法分离100 nt oligo(A)合成RNA寡核苷酸标准品(黑色色谱图)、核糖核酸酶T1酶解的EPO mRNA(蓝色色谱图)和核糖核酸酶T1酶解的Fluc Beta mRNA(红色色谱图)的结果,使用ACQUITY Premier BEH寡核苷酸分析专用柱(300 Å, 1.7 µm)。EPO mRNA poly(A)尾系列峰中丰度最高的峰对应于长度为124 nt的物质。Fluc Beta mRNA poly(A)尾峰组中丰度最高的峰对应于长度为128 nt的物质。
图2.IP RP LC方法分离100 nt oligo(A)合成RNA寡核苷酸标准品(黑色色谱图)、核糖核酸酶T1酶解的EPO mRNA(蓝色色谱图)和核糖核酸酶T1酶解的Fluc Beta mRNA(红色色谱图)的结果,使用ACQUITY Premier BEH寡核苷酸分析专用柱(300 Å, 1.7 µm)。EPO mRNA poly(A)尾系列峰中丰度最高的峰对应于长度为124 nt的物质。Fluc Beta mRNA poly(A)尾峰组中丰度最高的峰对应于长度为128 nt的物质。
所开发的IP RP LC方法不与MS检测严格兼容。使用100 nt寡核糖核苷酸腺苷标准品(黑色色谱图,图2),根据保留时间估计丰度最高的poly(A)寡核苷酸的长度。合成的100 nt寡核糖核苷酸(A)包含从全长产物中分离出来的短链n-x杂质。我们对图2中标记的80、85、90、95、99、100和101 nt物质绘制了线性趋势L(nt) = a × tr + b。L(nt)相对于保留时间tr的线性校正曲线如图3所示。利用该趋势估计在EPO mRNA和Fluc Beta mRNA酶解物中发现的主要poly(A)尾峰的长度L(nt)(比较图2和图3)。由于核糖核酸酶T1酶解物在G位置后裂解,因此释放的poly(A)寡核苷酸的5'端可能包含一个或几个额外的C、A或U核苷酸。Fluc Beta RNA poly(A)尾包含额外的C核苷酸正是这种情况。TriLink制造商未提供EPO mRNA的完整序列。要准确分配poly(A)尾物质的长度,必须考虑mRNA序列。需要时,还可以用核糖核酸酶A或其他特定核糖核酸酶对核糖核酸酶T1的酶解加以补充。
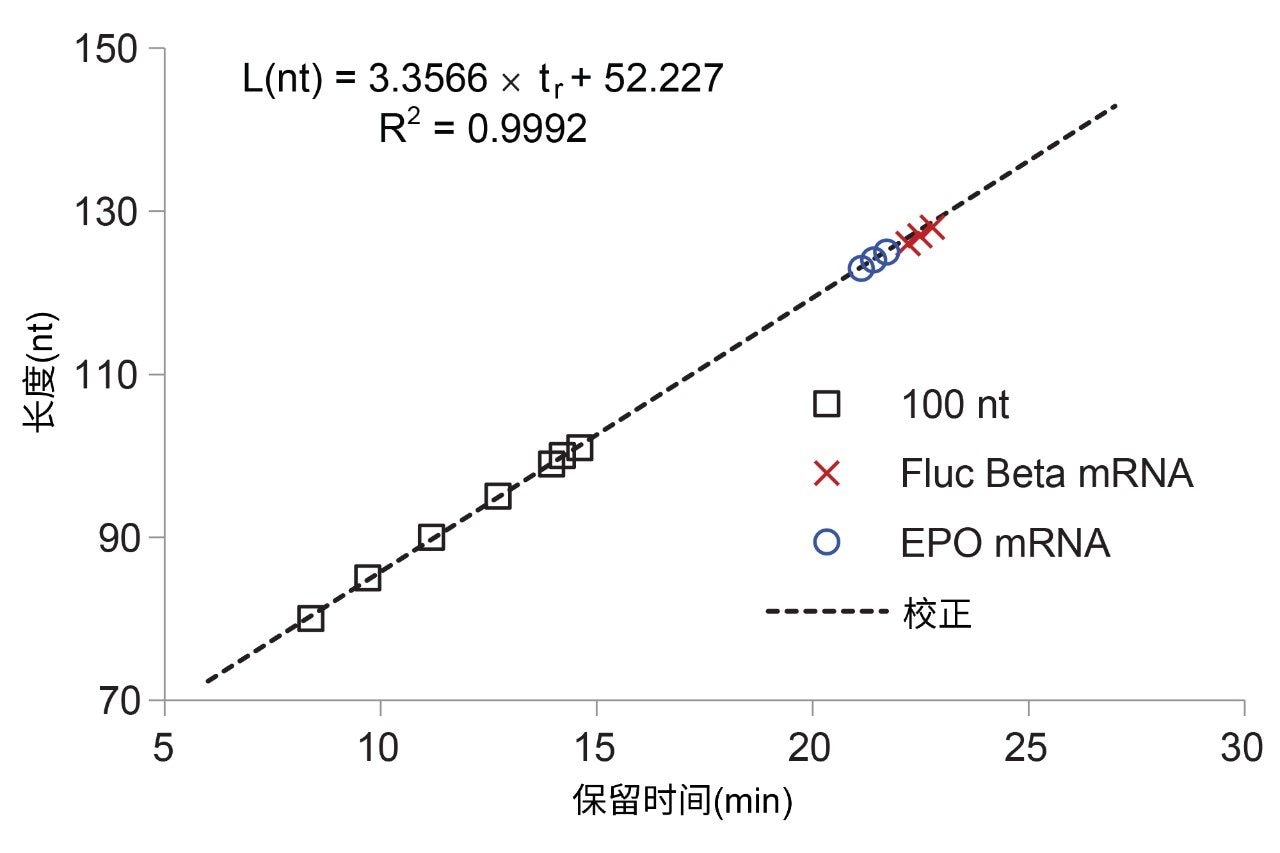 图3.使用100 nt合成RNA腺苷标准品及其截断的合成序列(图2中的黑色色谱图;80、85、90、95、99、100和101 nt峰)建立校正曲线L(nt) = a × tr + b。根据该校正曲线计算mRNA样品中最主要的poly(A)尾峰的长度。EPO mRNA和Fluc Beta mRNA酶解物主要的poly(A)尾峰分别为124 nt和128 nt。为使图更清晰,图3中仅绘制了在图2中检测到的三个丰度最高的峰。
图3.使用100 nt合成RNA腺苷标准品及其截断的合成序列(图2中的黑色色谱图;80、85、90、95、99、100和101 nt峰)建立校正曲线L(nt) = a × tr + b。根据该校正曲线计算mRNA样品中最主要的poly(A)尾峰的长度。EPO mRNA和Fluc Beta mRNA酶解物主要的poly(A)尾峰分别为124 nt和128 nt。为使图更清晰,图3中仅绘制了在图2中检测到的三个丰度最高的峰。
为确认这两个poly(A)尾样品中丰度最高的物质的分配结果,我们还单独使用二异丙基乙胺/HFIP离子对流动相进行了LC-MS实验,并使用SEC测量了poly(A)尾(如应用纪要720007853所述)。这三种方法(IP RP LC UV、LC-MS和SEC)对丰度最高的poly(A)尾峰都提供了一致的长度测量结果,即,EPO mRNA和Fluc Beta mRNA酶解物分别为124 nt ± 1 nt和128 nt ± 1 nt。
结论
本研究开发出一种分析mRNA poly(A)尾异质性的IP RP LC-UV方法。优化后的方法使用了大孔径ACQUITY Premier BEH C18寡核苷酸分析专用柱(300 Å, 150 × 2.1 mm, 1.7 mm)和高效的乙酸辛铵离子对缓冲液。长达150 nt的长链寡核苷酸也获得了理想的分离度,足以用于研究poly(A)尾样品的长度和异质性。所开发的方法使用简单而稳定的UV检测,适合用作质量控制分析。使用合成寡核糖核苷酸(A)标准品进行校正,来分配主要poly(A)尾寡核苷酸的长度。本研究的ACQUITY Premier UPLC系统和色谱柱采用了MaxPeak™高性能表面,可确保寡核苷酸分析物获得可重现的高回收率[4.15–17]。
参考资料
- M. Donegan, J.M. Nguyen, and M. Gilar, J. Chromatogr.A, 1666, 462860 (2022).DOI: 10.1016/j.chroma.2022.462860.
- M. Gilar, K.J. Fountain, Y. Budman, U.D. Neue, K.R. Yardley, P.D. Rainville, R.J. Russell, 2nd, and J.C. Gebler, J. Chromatogr.A, 958, 167–182 (2002).
- S.G. Roussis, M. Pearce, and C. Rentel, J. Chromatogr.A, 1594, 105–111 (2019).DOI: 10.1016/j.chroma.2019.02.026.
- G.J. Guimaraes, J.M. Sutton, M. Gilar, M. Donegan, and M.G. Bartlett, J. Pharm.Biomed.Anal., 208, 114439 (2022).DOI: 10.1016/j.jpba.2021.114439.
- A. Goyon, P. Yehl, and K. Zhang, J. Pharm.Biomed.Anal., 182, 113105 (2020).DOI: 10.1016/j.jpba.2020.113105.
- T. Jiang, N. Yu, J. Kim, J.R. Murgo, M. Kissai, K. Ravichandran, E.J. Miracco, V. Presnyak, and S. Hua, Anal.Chem., 91, 8500–8506 (2019).DOI: 10.1021/acs.analchem.9b01664.
- V.B. Ivleva, Y.Q. Yu, and M. Gilar, Rapid Commun Mass Spectrom, 24, 2631–2640 (2010).DOI: 10.1002/rcm.4683.
- L.T. França, E. Carrilho, and T.B. Kist, Q. Rev. Biophys., 35, 169–200 (2002).DOI: 10.1017/s0033583502003797.
- E. Carrilho, M.C. Ruiz-Martinez, J. Berka, I. Smirnov, W. Goetzinger, A.W. Miller, D. Brady, and B.L. Karger, Anal.Chem., 68, 3305–3313.(1996).
- M. Gilar, A. Belenky, Y. Budman, D.L. Smisek, and A.S. Cohen, J Chromatogr B Biomed Sci Appl, 714, 13–20 (1998).
- J.R. Thayer, Thermo Scientific Application note 21996, (2014).
- C.G. Huber, E. Stimpf, P.J. Oefner, and G.K. Bonn, LC-GC, 14, 114–127 (1996).
- M. Gilar and U.D. Neue, J. Chromatogr.A, 1169, 139–150 (2007).DOI: 10.1016/j.chroma.2007.09.005.
- S.M. McCarthy, M. Gilar, and J. Gebler, Anal Biochem, 390, 181-188 (2009).DOI: 10.1016/j.ab.2009.03.042.
- J.M. Nguyen, M. Gilar, B. Koshel, M. Donegan, J. MacLean, Z. Li, and M.A. Lauber, Bioanalysis, (2021).DOI: 10.4155/bio-2021–0115.
- M. Gilar, M. DeLano, and F. Gritti, J. Chromatogr.A, 1650, 462247 (2021).DOI: 10.1016/j.chroma.2021.462247.
- M. DeLano, T.H. Walter, M.A. Lauber, M. Gilar, M.C. Jung, J.M. Nguyen, C. Boissel, A.V. Patel, A. Bates-Harrison, and K.D. Wyndham, Anal.Chem., 93, 5773–5781 (2021).DOI: 10.1021/acs.analchem.0c05203.
720007873ZH,2023年2月