CONFIRM Sequence:一款用于合成寡核苷酸及其杂质测序的waters_connect™应用程序
摘要
本应用纪要展示了一套用于寡核苷酸及其杂质序列确认的自动化、合规的液质联用(LC-MS)工作流程。
优势
- CONFIRM Sequence是沃特世开发出的一款新的waters_connect应用程序,能够在寡核苷酸分析中快速解析复杂的串联质谱(MS/MS)和MSE(不选择特定母离子)质谱。
- CONFIRM Sequence应用程序对全长产物(FLP)以及低丰度寡核苷酸杂质的序列覆盖率可达100%,根据UV峰面积测量,可测序的丰度水平低至0.2%
- CONFIRM Sequence应用程序能够快速解析复杂寡核苷酸的碎片离子谱图数据
- 使用适当的碰撞能量梯度进行数据非依赖型采集(MSE或DIA)可对FLP提供100%序列覆盖率,而无需对单个寡核苷酸母离子进行碰撞能量优化
简介
合成寡核苷酸是近十年来出现的一类治疗药物,可替代小分子和蛋白治疗药物1-3。 寡核苷酸类药物的生产和质量控制需要使用高选择性、高灵敏度的LC-MS方法。离子对反相色谱法(IP-RP)是表征合成寡核苷酸的常用LC-MS方法,尽管固相寡聚填料取得了重大进展4,但合成寡核苷酸中仍含有多种微量(0.1–2%)杂质3-5。
此前已有文献报道过使用BioAccord™系统在符合法规要求的环境中对合成寡核苷酸及其杂质进行完整质量数确认的自动化工作流程6–9。除完整质量数确认外,全面表征寡核苷酸的另一个基石是序列确证/验证过程。序列准确性是决定寡核苷酸治疗药物和其他核酸类药物(例如mRNA)活性的重要因素,因为它们的核苷酸序列与其在细胞内的生物学功能直接相关。要实现明确地序列匹配,需要序列覆盖率达到100%。在通过串联质谱法对寡核苷酸母离子进行气相碎裂后,分析所有检测到的碎片离子以确认预期的寡核苷酸序列。如果手动执行此过程,可能会非常乏味、耗时,因为寡核苷酸碎裂的可能性很多,通常会在寡核苷酸母离子发生碰撞诱导解离(CID)后观察到种类繁多的a、b、c、d、w、x、y和z离子10。由于碎片离子的多样性,再加上序列无信息片段的存在(例如核酸碱基的丢失),寡核苷酸的明确测序显然比肽测序更具挑战性。
在过去的二十年里,人们已经开发出几种计算机程序来自动注释这种复杂的气相解离谱图11-18。
CONFIRM Sequence是最近推出的一款waters_connect应用程序,可通过自动处理由靶向MS/MS或非靶向MSE (DIA)获得的寡核苷酸的MS/MS谱图,对合成寡核苷酸及其杂质进行自动测序。
在本研究中,我们考察了CONFIRM Sequence应用程序对合成寡核苷酸及其杂质进行快速、自动测序的能力。
实验
试剂和样品前处理
三乙胺(TEA,纯度99.5%,产品目录号65897-50ML)和甲醇(LC-MS级,产品目录号34966-1L)购自Honeywell(美国卡罗来纳州夏洛特),1,1,1,3,3,3-六氟-2-丙醇(HFIP,纯度99%,产品目录号105228-100G)购自Sigma Aldrich(美国密苏里州圣路易斯)。HPLC级去离子(DI) I级水使用MilliQ系统(密理博公司,美国马萨诸塞州贝德福德)净化。流动相现配现用。21 mer高度修饰的寡核苷酸,在其19个核苷上含有2'-OMe修饰,序列为GUA ACC AAG AGU AUU CCA UTT,元素组成为C229H306N76O143P20,购自ATDBio(英国南安普敦)。用去离子水配制浓度为1 µM(或2.34 µg/mL)的储备液,进样体积10 µL,相当于柱上进样10 pmol的21 mer寡核苷酸用于MS/MS采集。
液相色谱条件
|
LC-MS系统: |
Xevo™ G2-XS QTof与H-Class Bio UPLC™联用系统 |
|
色谱柱: |
ACQUITY™ Premier OST色谱柱 1.7 µm, 130 Å, 2.1 x 100 mm, (P/N: 186009485)
|
|
柱温: |
60 °C |
|
流速: |
300 µL/min |
|
流动相 |
|
|
溶剂A: |
40 mM HFIP(六氟异丙醇),7 mM TEA(三乙胺),溶于去离子水中,pH 8.6 |
|
溶剂B: |
20 mM HFIP,3.5 mM TEA,溶于50%甲醇 |
|
样品温度: |
6 °C |
|
样品瓶: |
LC-MS认证的最大回收样品瓶(P/N: 186005663CV) |
|
进样体积: |
10 µL |
|
清洗溶剂 |
|
|
清除溶剂: |
50%甲醇 |
|
样品管理器清洗溶剂: |
50%甲醇 |
|
密封清洗液: |
20%乙腈的去离子水溶液 |
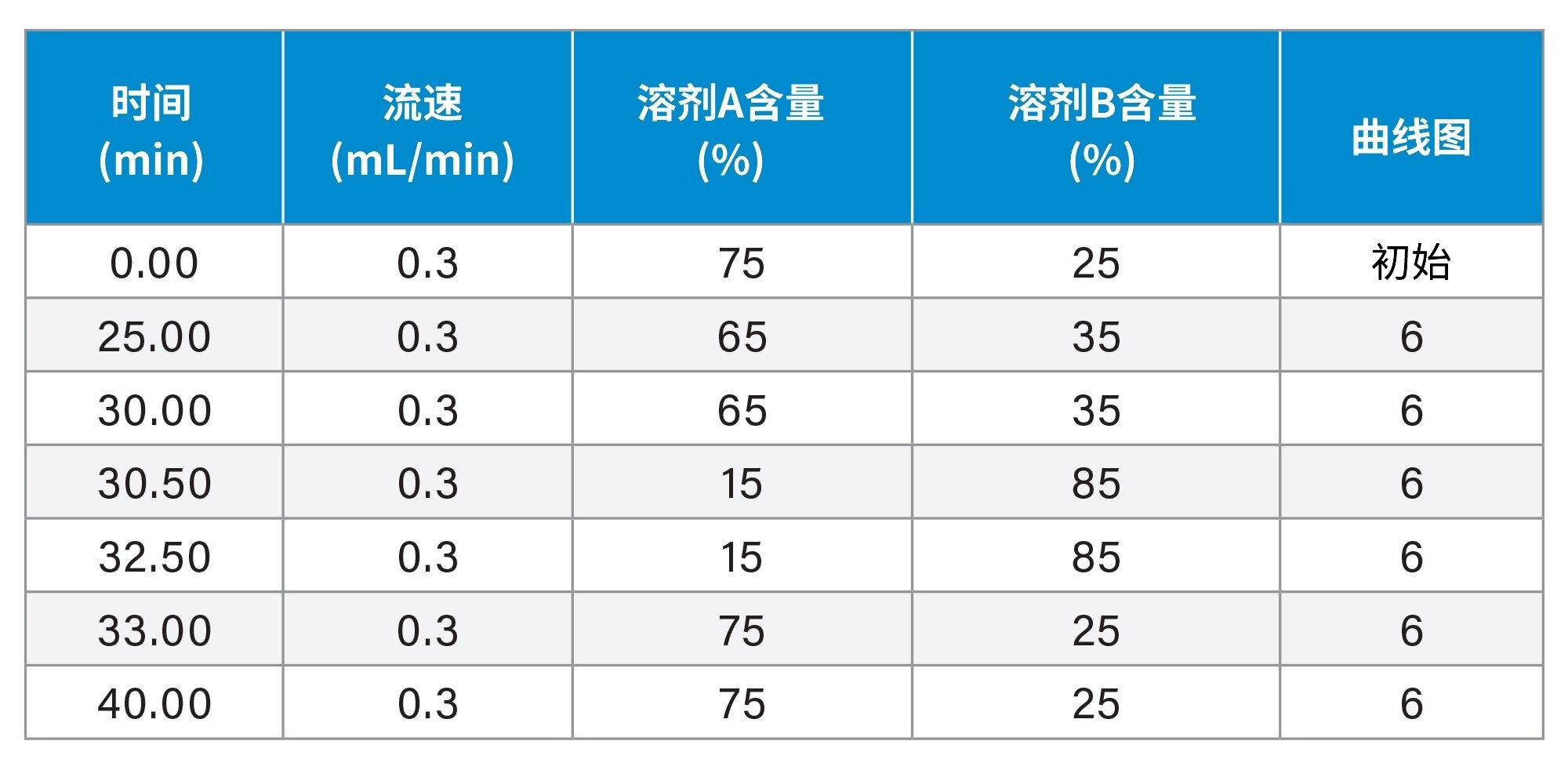
梯度表
质谱条件
|
毛细管电压: |
2.5 kV |
|
锥孔电压: |
40 V |
|
离子源温度: |
125 °C |
|
脱溶剂气温度: |
400 °C |
|
脱溶剂气(N2)流速: |
600 L/h |
|
锥孔气流速: |
50 L/h |
|
TOF质量范围: |
500-5000 |
|
采集速率: |
1 Hz |
|
碰撞能量: |
针对靶向MS/MS采集双电荷、三电荷和四电荷寡核苷酸母离子研究了5~70 V范围内的15个固定碰撞能量(2、5、10、15、20、25、30、35、40、45、50、55、60、65和70 V);还针对MSE(DIA)采集研究了几个高能量CE梯度(20~40、40~60、60~80和80~100 V) |
|
实时校正标准液: |
100 fmol/µL GFP |
|
数据采集软件: |
waters_connect 1.9.13.9 |
|
数据处理软件: |
CONFIRM Sequence 1.0 |
结果与讨论
选择21-mer寡核苷酸及其杂质作为测试样品来展示新推出的CONFIRM Sequence应用程序的性能和功能。寡核苷酸基本由经过修饰的核苷酸构成(21个核苷酸中有19个经过修饰),如实验部分和图1中列出的序列所示。在寡核苷酸序列中,2'-OMe修饰连接到三个蓝色标记的鸟苷(G)和七个绿色标记的腺苷(A)上。除了将相同的2'-OMe官能团连接到尿苷和胞苷外,这两个核苷的核碱基上还连接了5-Me进一步修饰,产生五个2'-OMe 5-Me尿苷(U,紫色标记)和四个2'-OMe 5-Me胞苷(C,红色标记)。唯一未修饰的核苷酸是21 mer 3'端的两个脱氧胸苷(TT)。
CONFIRM Sequence应用程序的一个重要组件是合成库,用户可以使用合成库定义目标寡核苷酸序列。该库分为四个独立的库:亚成分库、单体库、序列库和修饰物库。所有的核苷酸单体都可以从结构上分为三个单独的分子实体:碱基、糖和连接基团,它们各自分别保存在亚成分库中。这些核苷酸构件组装成单体,单独保存在单体库中。用户可以将多种核苷酸单体组合成所需的寡核苷酸序列,该序列保存在序列库中,序列库中的序列在需要时可以轻松编辑。如果寡核苷酸包含非标准(异常)核苷酸修饰,则可以从修饰物库部分手动配置这些修饰。对于本研究所分析的21-mer高度修饰寡核苷酸,在单体库中配置了两个单体,因为2'-OMe-5-Me胞苷和2'-OMe-5-Me尿苷不属于库内的默认内容。图1所示的屏幕截图显示了使用相应亚成分组装这两个核苷酸单体的具体操作。在CONFIRM Sequence应用程序中,高度修饰的21-mer寡核苷酸的序列输入为:OMEG 2-OMe-5-MeU OMEA OMEA 2-OMe-5-MeC 2-OMe-5-MeC OMEA OMEA OMEG OMEA OMEG 2-OMe-5-MeU OMEA 2-OMe-5-MeU 2-OMe-5-MeU 2-OMe-5-MeC 2-OMe-5-MeC OMEA 2-OMe-5-MeU dT dT。
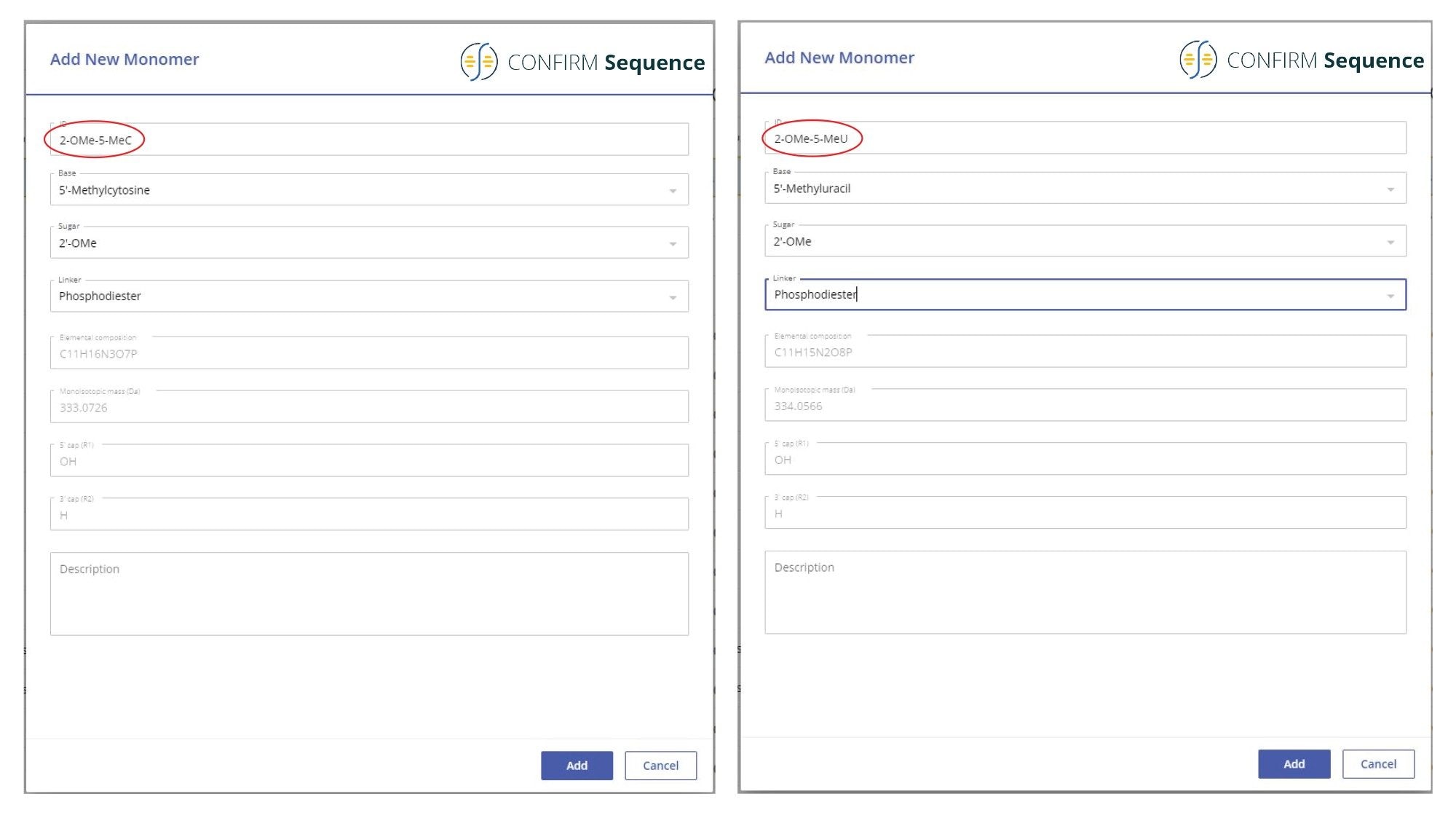 图1.单体库的屏幕截图,详细说明了使用相应的单个亚成分(碱基、糖和连接基团)组装两个核苷酸单体(2'-OMe-5-Me胞苷和2'-OMe-5-Me尿苷)的具体操作。
图1.单体库的屏幕截图,详细说明了使用相应的单个亚成分(碱基、糖和连接基团)组装两个核苷酸单体(2'-OMe-5-Me胞苷和2'-OMe-5-Me尿苷)的具体操作。
利用2.1 x 100 mm ACQUITY Premier OST色谱柱(P/N: 186009485)分析21nt寡核苷酸时,分离结果显示出相当复杂的杂质分布,如图2所示。TUV和MS检测器共分离和检测出11种寡核苷酸杂质。第一次进样时,进样2 µL的1 µM 21-mer寡核苷酸,并在负离子ESI全扫描模式(m/z=500–5000)下采集数据,以找到该寡核苷酸及其杂质对应的最高丰度母离子(见表I)。在连续进样中,进样10 µL样品,在Xevo G2-XS仪器的MS/MS模式下使用四极杆分离(~5 Da窗口)某些多电荷寡核苷酸母离子(见表I)。经过四极杆分离之后,每个母离子在仪器的碰撞室内发生CID(碰撞诱导解离)碎裂,产生包含相应碎片离子的MS/MS谱图。在10-80 V的施加电压范围内进行了碰撞能量研究,以找出使每个寡核苷酸产生最佳碎裂(更高的碎片离子序列覆盖率)的最佳固定碰撞电压。CONFIRM Sequence应用程序用于根据记录的所有MS/MS谱图来评估MS/MS序列覆盖率。对于本例中的21-mer寡核苷酸,当它的三电荷母离子([M-3H]-3=2341.45)在63 V的固定碰撞电压下发生碎裂时,序列覆盖率达到100%。CONFIRM Sequence应用程序采取点图可视化显示,可以轻松评估预测序列的覆盖率,如图3中的屏幕截图所示。根据该图,按照寡核苷酸碎裂命名法检测并匹配了多种碎片离子10。在数据分析方面,CONFIRM Sequence使用内部生成的靶向同位素簇算法将预测的寡核苷酸碎片与在原始数据中检测到的碎片进行匹配。软件将显示相关的匹配信息(以图形和表格形式),并提供每个匹配碎片的统计分析。使用点图显示有利于轻松评估预测序列的覆盖率,或定位杂质修饰。用户还可以使用CONFIRM Sequence应用程序检查原始MS/MS碎裂谱图,如图4所示。该谱图中的大部分离子信号在图中显示为绿色,表明它们已与预测的碎片离子匹配。当使用MSE采集分析样品时,相同的寡核苷酸获得了相同的最大序列覆盖率(100%)。本例通过先用低碰撞能量检测母离子(10 V),再以碰撞能量梯度(40~60 V)诱导CID同时碎裂所有母离子,两种MS扫描模式交替1秒的方式,进行数据非依赖型采集。图5中显示的CONFIRM Sequence应用程序的点图结果表明,MSE采集可以在单次进样中产生高质量碎片离子数据,且无需优化碰撞能量。如图6的处理结果所示,在BioAccord仪器(小型台式TOF MS系统)上使用相同类型的数据采集(MSE)可以获得相对较高的序列覆盖率(>70%)。与图3和图5中所示的数据相比,BioAccord采集的序列覆盖率较低,这是因为几个低m/z碎片离子超出了m/z=400–5000 Da的采集质量数范围。
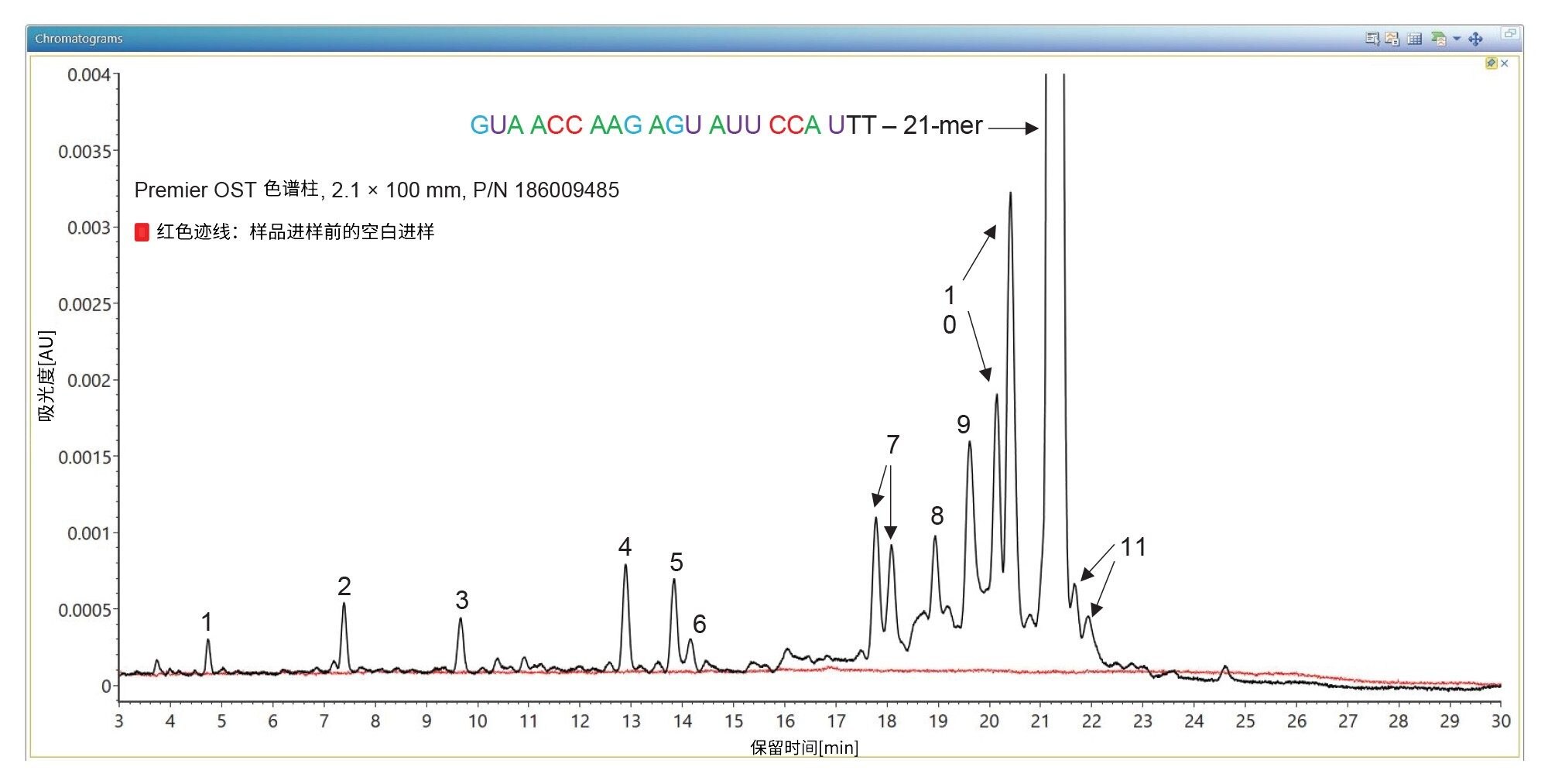 图2.21-mer高度修饰的寡核苷酸与其杂质分离结果的TUV色谱图。Premier OST色谱柱可分离杂乱的寡核苷酸序列(如峰7和峰10对应的双峰)和全长产物的脱氨基异构体(峰11)。
图2.21-mer高度修饰的寡核苷酸与其杂质分离结果的TUV色谱图。Premier OST色谱柱可分离杂乱的寡核苷酸序列(如峰7和峰10对应的双峰)和全长产物的脱氨基异构体(峰11)。
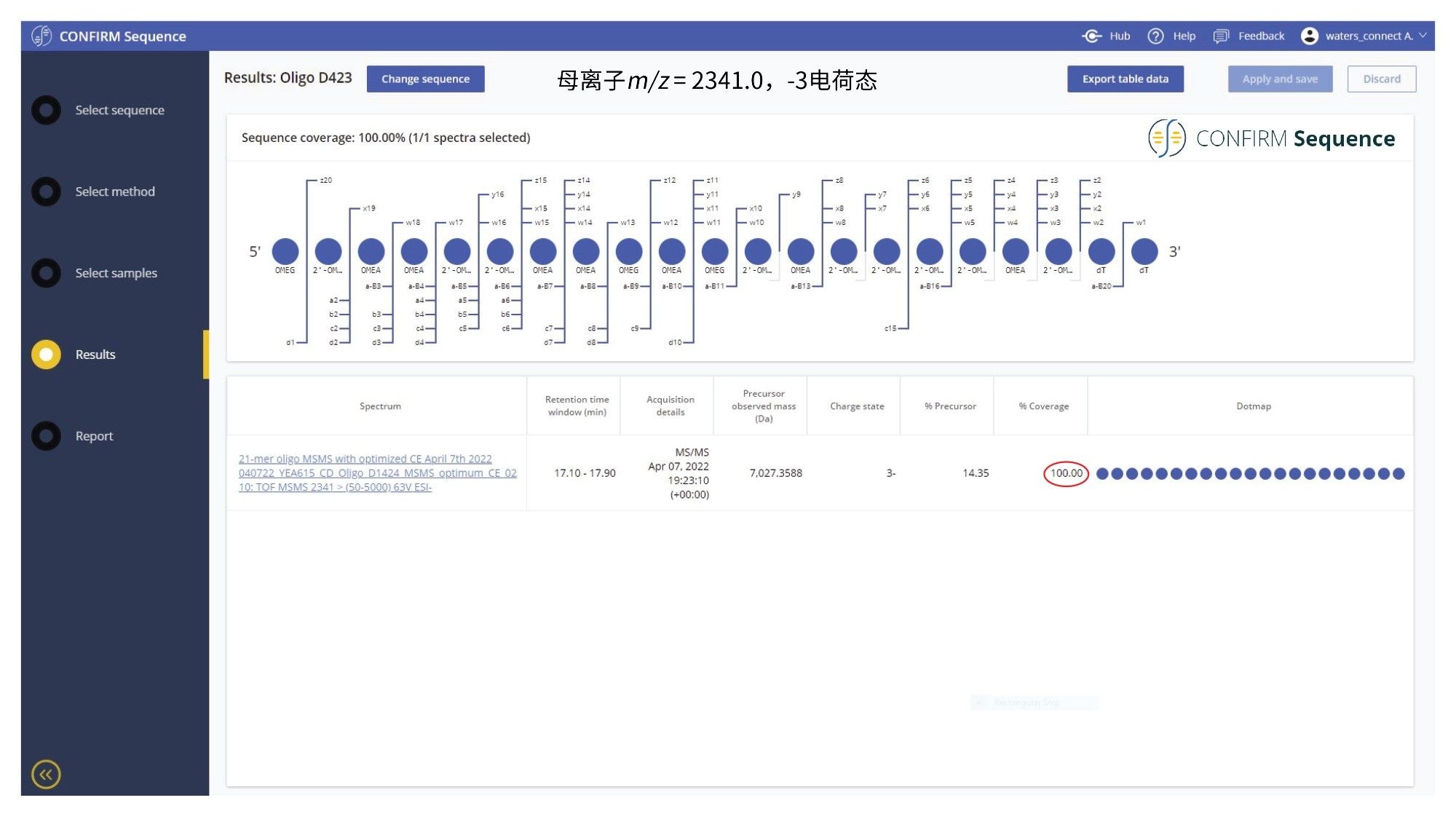 图3.CONFIRM Sequence屏幕截图,以点图形式展示了优异的MS/MS碎裂覆盖率(100%)。在Xevo G2-XS QTof仪器的碰撞室内使用最佳固定碰撞能量(设置为63 V)对21-mer高度修饰寡核苷酸(m/z = 2342.0)的[M-3H]-3 母离子进行碎裂。
图3.CONFIRM Sequence屏幕截图,以点图形式展示了优异的MS/MS碎裂覆盖率(100%)。在Xevo G2-XS QTof仪器的碰撞室内使用最佳固定碰撞能量(设置为63 V)对21-mer高度修饰寡核苷酸(m/z = 2342.0)的[M-3H]-3 母离子进行碎裂。
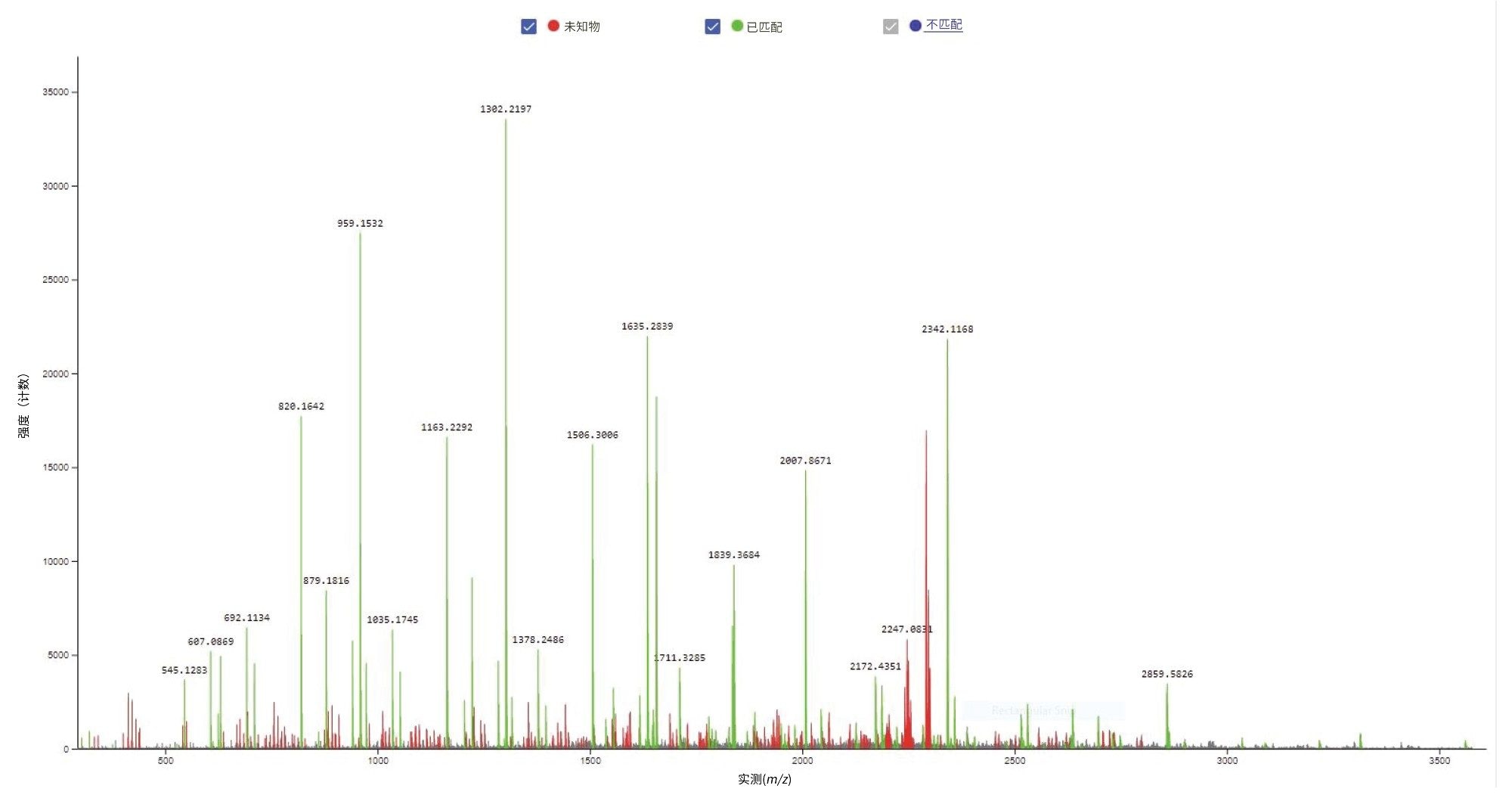 图4.CONFIRM Sequence屏幕截图,显示了21-mer寡核苷酸的MS/MS谱图记录。在Xevo G2-XS QTof仪器的碰撞室内使用最佳固定碰撞能量(设置为63 V)对该寡核苷酸(m/z = 2341.0)的[M-3H]-3 母离子进行碎裂。根据图3所示的点图,绿色标记的碎片离子已匹配到寡核苷酸序列。
图4.CONFIRM Sequence屏幕截图,显示了21-mer寡核苷酸的MS/MS谱图记录。在Xevo G2-XS QTof仪器的碰撞室内使用最佳固定碰撞能量(设置为63 V)对该寡核苷酸(m/z = 2341.0)的[M-3H]-3 母离子进行碎裂。根据图3所示的点图,绿色标记的碎片离子已匹配到寡核苷酸序列。
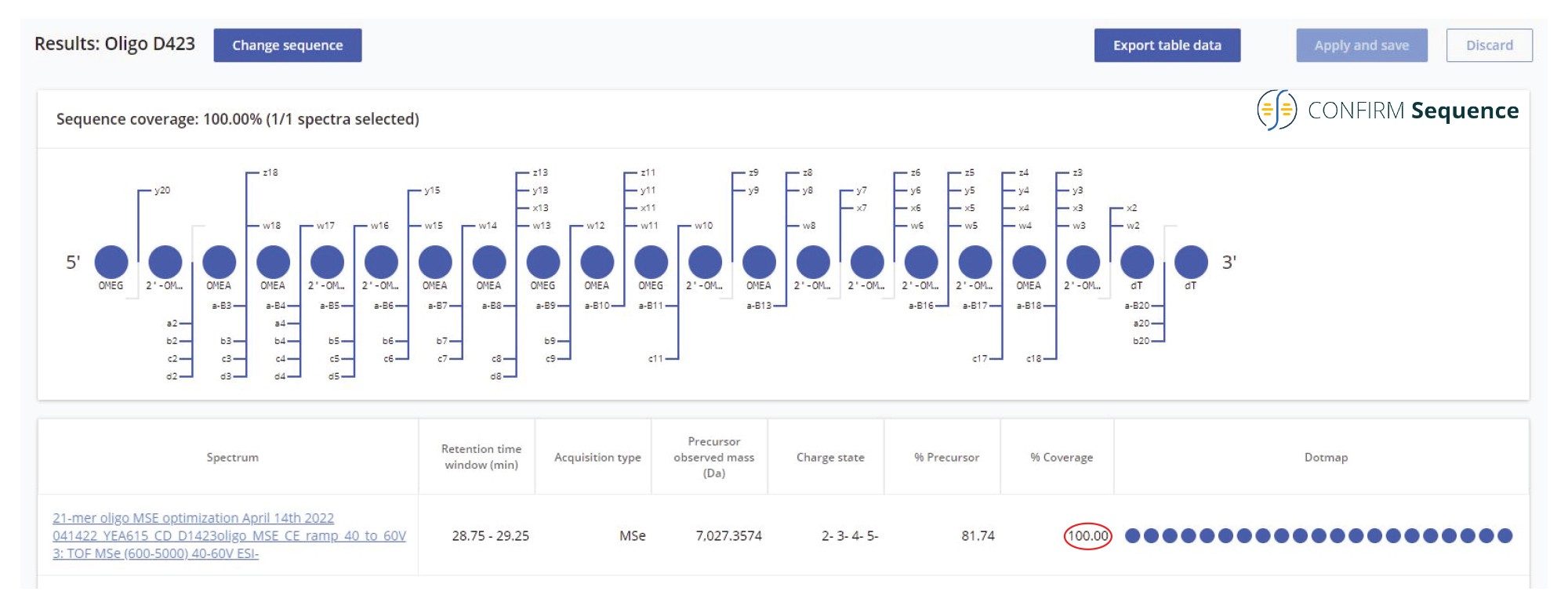 图5.以点图形式呈现的100%序列覆盖率,通过高能量MSE非靶向碎裂产生。在Xevo G2-XS仪器的碰撞室内施加最佳碰撞能量梯度(40~60 V),对21-mer高度修饰寡核苷酸的所有母离子进行碎裂。
图5.以点图形式呈现的100%序列覆盖率,通过高能量MSE非靶向碎裂产生。在Xevo G2-XS仪器的碰撞室内施加最佳碰撞能量梯度(40~60 V),对21-mer高度修饰寡核苷酸的所有母离子进行碎裂。
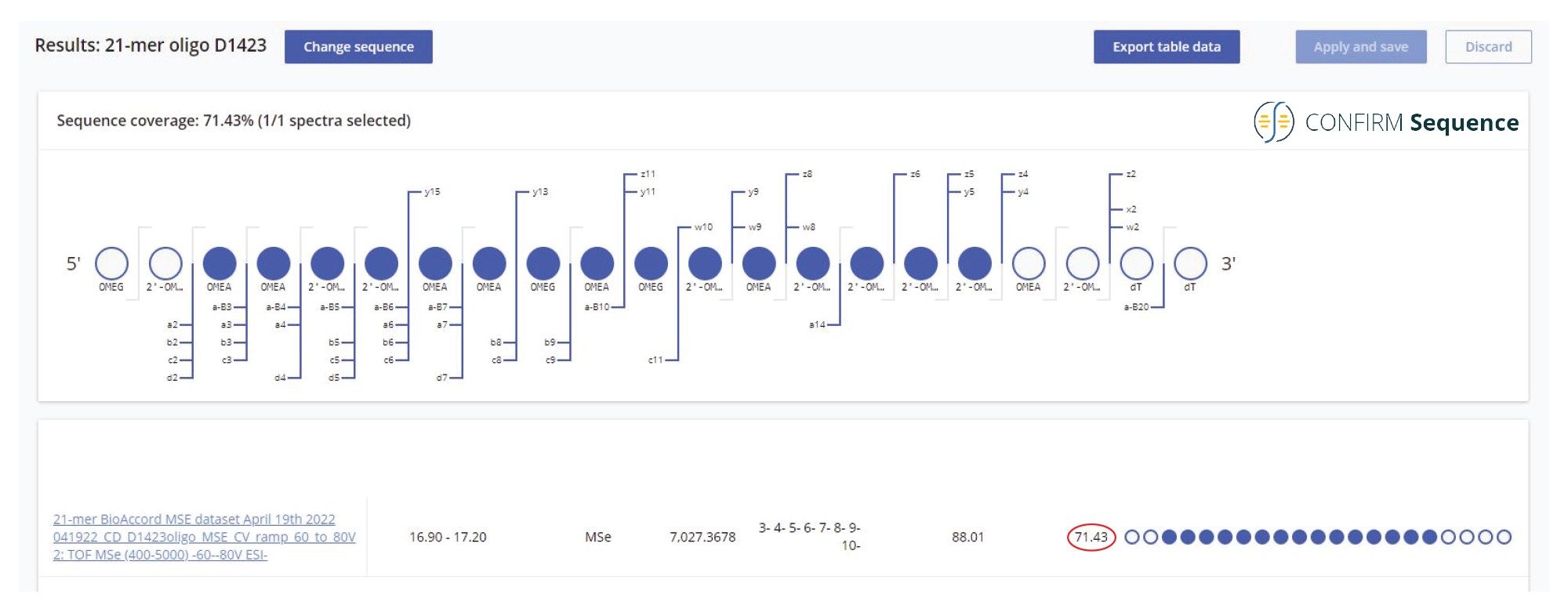 图6.在BioAccord上进行高能量MSE非靶向碎裂获得的高序列覆盖率(> 70%)。在BioAccord Tof仪器的Step Wave中施加最佳锥孔电压梯度(60~80 V),对21-mer高度修饰寡核苷酸的所有母离子进行碎裂。
图6.在BioAccord上进行高能量MSE非靶向碎裂获得的高序列覆盖率(> 70%)。在BioAccord Tof仪器的Step Wave中施加最佳锥孔电压梯度(60~80 V),对21-mer高度修饰寡核苷酸的所有母离子进行碎裂。
在Xevo G2-XS仪器上使用固定碰撞能量对全长产物以及七种微量杂质进行了测序,所用的碰撞能量已根据表I中给出的值针对每种寡核苷酸母离子进行了优化。
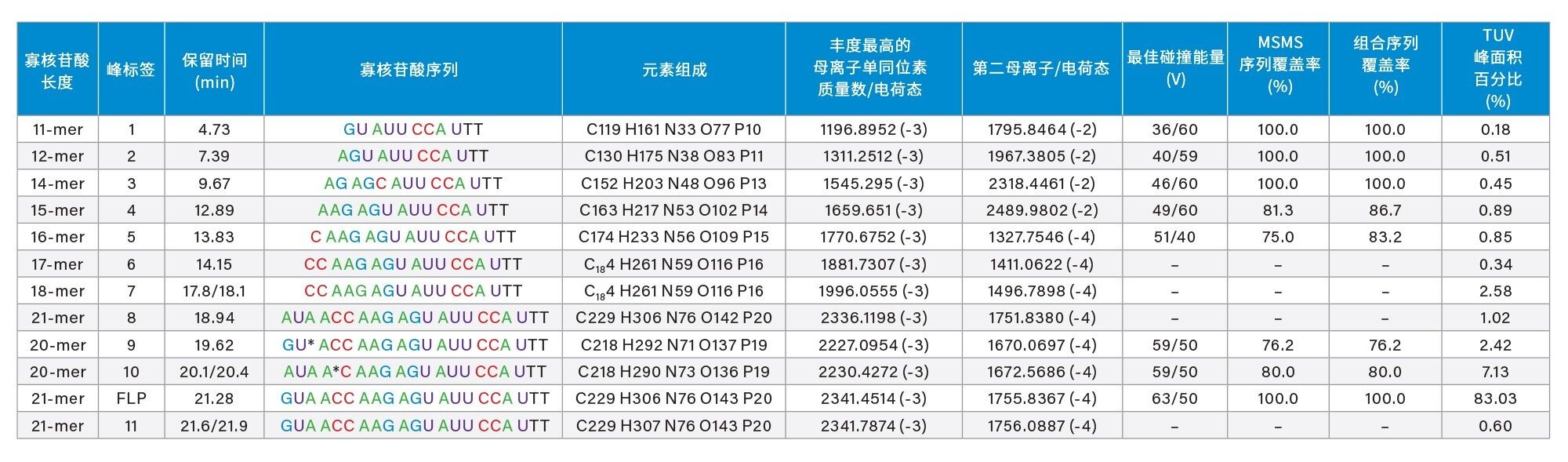 表I. 在经过大量修饰的21 mer寡核苷酸中鉴定出的11种寡核苷酸杂质。使用Xevo G2-XS仪器对八种杂质和全长产物(FLP)进行测序,并使用CONFIRM Sequence应用程序处理在最佳碰撞能量下碎裂得到的单个MS/MS谱图。FLP及其杂质的MS/MS序列覆盖率均高于75%。丰度最低的杂质,即11-mer寡核苷酸得到完全测序(100%序列覆盖率),FLP(21-mer寡核苷酸)的序列也确认获得100%覆盖率。总序列覆盖率对应于每种寡核苷酸杂质的两个母离子的MS/MS碎裂获得的组合序列。
表I. 在经过大量修饰的21 mer寡核苷酸中鉴定出的11种寡核苷酸杂质。使用Xevo G2-XS仪器对八种杂质和全长产物(FLP)进行测序,并使用CONFIRM Sequence应用程序处理在最佳碰撞能量下碎裂得到的单个MS/MS谱图。FLP及其杂质的MS/MS序列覆盖率均高于75%。丰度最低的杂质,即11-mer寡核苷酸得到完全测序(100%序列覆盖率),FLP(21-mer寡核苷酸)的序列也确认获得100%覆盖率。总序列覆盖率对应于每种寡核苷酸杂质的两个母离子的MS/MS碎裂获得的组合序列。
在该样品中检测到的最低丰度杂质对应于一个11-mer寡核苷酸,UV峰面积测量显示该杂质的含量为0.2%。如图2所示,该11-mer寡核苷酸是LC-UV色谱图中洗脱的第一个色谱峰,被标记为峰1(参见图1和表I)。该杂质的ESI-MS谱图表明,该寡核苷酸仅存在两个可用于MS/MS碎裂的母离子,如图7所示。使用39 V的固定碰撞电压对最高丰度的母离子,即m/z=1196.89处的三电荷[M-3H]-3离子进行碎裂,产生了包含大量碎片的MS/MS谱图。如图8中的CONFIRM Sequence结果所示,这些种类繁多的碎片离子覆盖了该寡核苷酸杂质的100%序列。图3和图8中显示的数据表明,当在Xevo G2-XS QTof仪器上进行测序时,无论是高丰度寡核苷酸(例如FLP 21-mer),还是丰度水平低至0.2%(11-mer杂质)的低丰度寡核苷酸杂质,都可以得到100%序列覆盖率的结果。通过CONFIRM Sequence应用程序验证发现,11-mer寡核苷酸杂质以及其他四种杂质(标记为峰2-5)均对应于21-mer FLP的截短版本,即在母体寡核苷酸的5’端缺失一串长5-10个核苷酸的序列。这些匹配失败的序列是I类杂质5,通常作为3'端到5'端寡核苷酸合成的副产物存在3,4。CONFIRM Sequence应用程序能够结合相同寡核苷酸杂质的多个母离子碎裂结果,以增加序列覆盖率。此方法提高了标记为峰4和5的杂质的序列覆盖率,如表I中的“组合序列覆盖率”列所示。
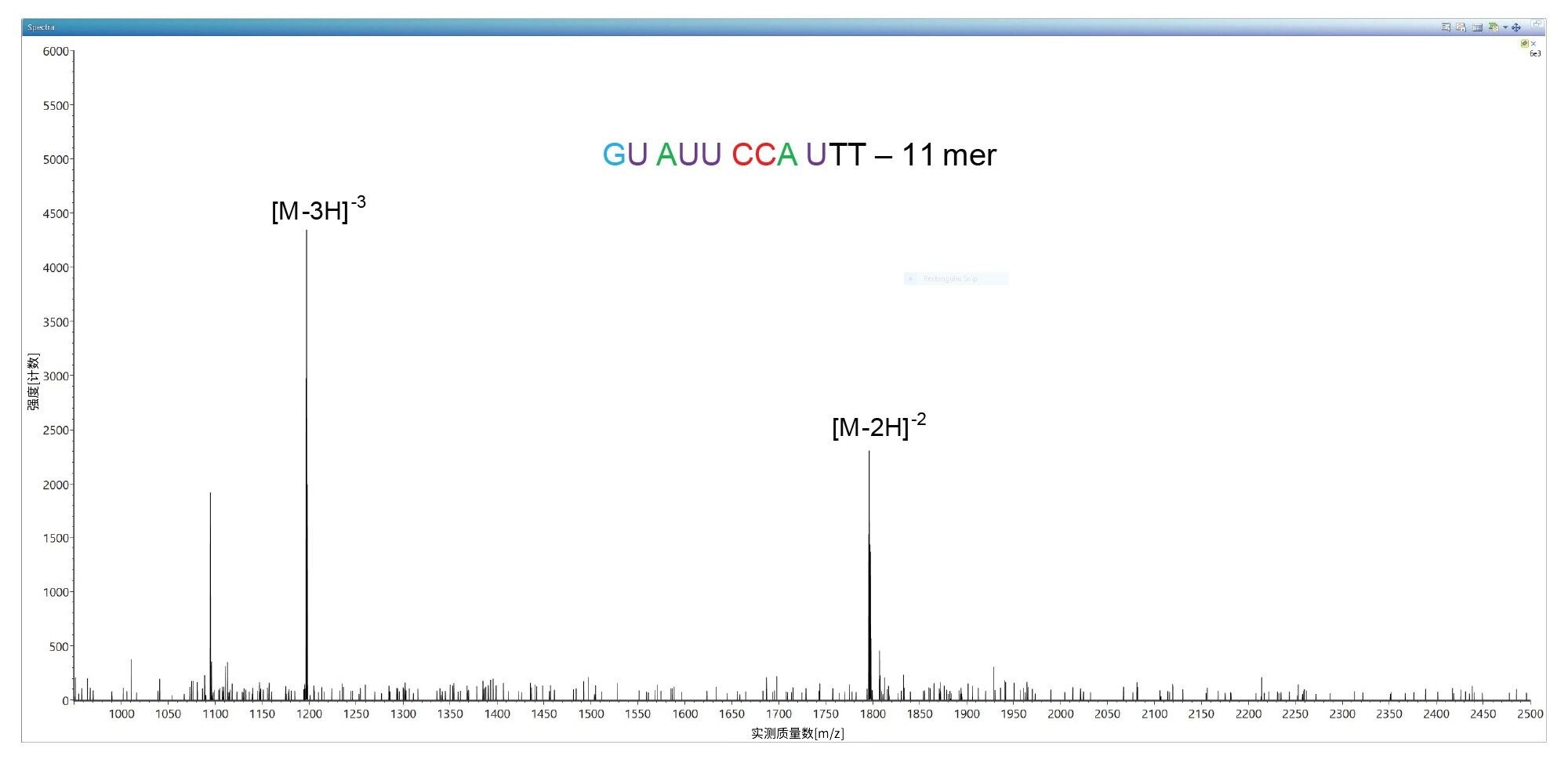 图7.11-mer寡核苷酸杂质的离子配对反相ESI-MS谱图,该杂质是21-mer寡核苷酸样品中丰度最低的杂质(相对丰度为0.2%,在图1的色谱图中标记为峰1)。
图7.11-mer寡核苷酸杂质的离子配对反相ESI-MS谱图,该杂质是21-mer寡核苷酸样品中丰度最低的杂质(相对丰度为0.2%,在图1的色谱图中标记为峰1)。
![MS/MS碎裂11-mer寡核苷酸杂质的[M-3H]-3母离子获得了100%序列覆盖率。在Xevo G2-XS仪器的碰撞室内使用最佳固定碰撞能量(36 V)对母离子进行碎裂](/content/dam/waters/zh/app-notes/2022/720007677/720007677en-f8.jpg.82.resize/img.jpg) 图8.MS/MS碎裂11-mer寡核苷酸杂质的[M-3H]-3母离子获得了100%序列覆盖率。在Xevo G2-XS仪器的碰撞室内使用最佳固定碰撞能量(36 V)对母离子进行碎裂。
图8.MS/MS碎裂11-mer寡核苷酸杂质的[M-3H]-3母离子获得了100%序列覆盖率。在Xevo G2-XS仪器的碰撞室内使用最佳固定碰撞能量(36 V)对母离子进行碎裂。
另一类寡核苷酸杂质是图2所示色谱图中标识为峰7、10和11的双峰。在本例中,每个双峰对应于一对具有相同(同量异位)母离子的寡核苷酸,因此这些杂质预计是源自母体分子的序列变体,属于III类杂质5。 最后一个洗脱的双峰(峰11)很可能对应于FLP的脱氨基形式,它们可能与21-mer序列中存在的2'-OMe-5-Me胞苷有关,因为这些胞苷易于发生脱氨基反应19。 如图2所示,ACQUITY Premier OST色谱柱能够分离寡核苷酸主要组件尾部如此小的修饰(+1 Da),这个结果十分令人瞩目。
21-mer寡核苷酸中丰度最高的杂质是两个序列变体20-mer,这种变体缺少一个2'-OMe-5-Me胞苷,在图2中标识为双峰10。21-mer序列中有4个经过修饰的胞苷,但由于它们成对存在,因此只有两种可能缺少一个2'-OMe-5 Me胞苷的20-mer杂质。CONFIRM Sequence应用程序可以搜索序列修饰(省略或添加),以尝试匹配已知序列和母离子质量数的MS/MS碎片离子。本例使用软件在21-mer序列中寻找缺失的2'-OMe-5 Me胞苷,并发现了强有力的证据(80%序列覆盖率)来分配此杂质的序列:GUA A*C AAG AGU AUU CCA UTT,第5位缺少核苷酸残基。与在第16位缺失相同残基的另一序列(序列覆盖率仅50%)相比,该寡核苷酸序列显示出显著更高的序列覆盖率(80%)。两种处理结果均已显示在图9中。图10显示了20-mer寡核苷酸对(属于图1中的峰10双峰并在FLP之前洗脱)的最高丰度序列变体的MS/MS谱图记录。在Xevo G2-XS QTof仪器的碰撞室内使用最佳固定碰撞能量(设置为59 V)对该寡核苷酸(m/z=2230.42)的三电荷母离子进行碎裂。在m/z=1506.29处检测到的a-B5碎片离子证实,在该寡核苷酸的5'端附近存在一个2'-OMe-5-Me胞苷,而在m/z=1656.77处存在的w10碎片离子表示另外两个2'-OMe-5-Me胞苷位于分子的另一端。通过此方法,图10中的MS/MS谱图证实了最高丰度杂质的序列为:GUA AC AAG AGU AUU CCA UTT。
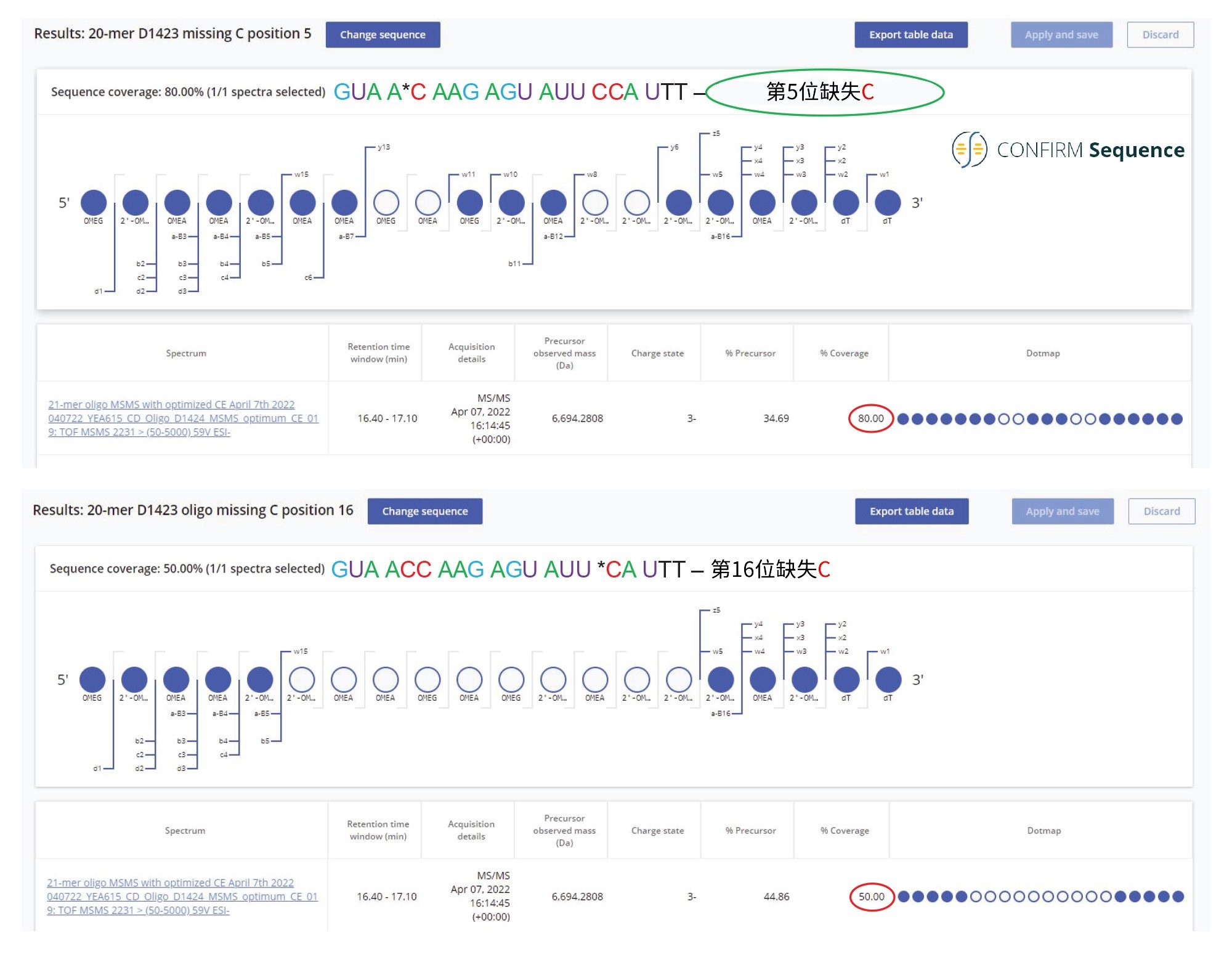 图9.CONFIRM Sequence屏幕截图,显示了图2中所示的双峰10所属20-mer寡核苷酸杂质的序列覆盖率。该杂质的最高丰度异构体在FLP之前洗脱,被鉴定为在第5位缺少2'-OMe 5 Me胞苷残基的寡核苷酸。与在第16位缺失相同残基的另一序列相比,该寡核苷酸序列显示出显著更高的序列覆盖率(80%)。21-mer序列中有4个经过修饰的胞苷,但由于它们成对存在,因此只有两种可能缺少一个2'-OMe5 Me胞苷的20-mer杂质。使用软件在21-mer序列中寻找缺失的2'-OMe-5-Me胞苷,并发现了强有力的证据(80%序列覆盖率)来分配此杂质的序列:GUA A*C AAG AGU AUU CCA UTT。
图9.CONFIRM Sequence屏幕截图,显示了图2中所示的双峰10所属20-mer寡核苷酸杂质的序列覆盖率。该杂质的最高丰度异构体在FLP之前洗脱,被鉴定为在第5位缺少2'-OMe 5 Me胞苷残基的寡核苷酸。与在第16位缺失相同残基的另一序列相比,该寡核苷酸序列显示出显著更高的序列覆盖率(80%)。21-mer序列中有4个经过修饰的胞苷,但由于它们成对存在,因此只有两种可能缺少一个2'-OMe5 Me胞苷的20-mer杂质。使用软件在21-mer序列中寻找缺失的2'-OMe-5-Me胞苷,并发现了强有力的证据(80%序列覆盖率)来分配此杂质的序列:GUA A*C AAG AGU AUU CCA UTT。
![MS/MS谱图记录了标记为峰10并在FLP之前洗脱的21-mer寡核苷酸杂质的最高丰度异构体。在Xevo G2-XS QTof仪器的碰撞室内使用最佳固定碰撞能量(设置为59 V)对该寡核苷酸(m/z = 2231.0)的[M-3H]-3 母离子进行碎裂。在m/z=1506.29处检测到的a-B5碎片离子证实,在该寡核苷酸的5'端附近存在一个2'-OMe-5-Me胞苷,而在m/z=1656.77处存在的w10碎片离子表示另外两个2'-OMe-5-Me胞苷位于分子的另一端](/content/dam/waters/zh/app-notes/2022/720007677/720007677en-f10.jpg.82.resize/img.jpg) 图10.MS/MS谱图记录了标记为峰10并在FLP之前洗脱的21-mer寡核苷酸杂质的最高丰度异构体。在Xevo G2-XS QTof仪器的碰撞室内使用最佳固定碰撞能量(设置为59 V)对该寡核苷酸(m/z = 2231.0)的[M-3H]-3 母离子进行碎裂。在m/z=1506.29处检测到的a-B5碎片离子证实,在该寡核苷酸的5'端附近存在一个2'-OMe-5-Me胞苷,而在m/z=1656.77处存在的w10碎片离子表示另外两个2'-OMe-5-Me胞苷位于分子的另一端。
图10.MS/MS谱图记录了标记为峰10并在FLP之前洗脱的21-mer寡核苷酸杂质的最高丰度异构体。在Xevo G2-XS QTof仪器的碰撞室内使用最佳固定碰撞能量(设置为59 V)对该寡核苷酸(m/z = 2231.0)的[M-3H]-3 母离子进行碎裂。在m/z=1506.29处检测到的a-B5碎片离子证实,在该寡核苷酸的5'端附近存在一个2'-OMe-5-Me胞苷,而在m/z=1656.77处存在的w10碎片离子表示另外两个2'-OMe-5-Me胞苷位于分子的另一端。
结论
- 沃特世推出了一款新的waters_connect应用程序 - CONFIRM Sequence,用于快速解析复杂的MS/MS和MSE (DIA)寡核苷酸质谱。
- CONFIRM Sequence应用程序对全长产物(FLP)以及低丰度寡核苷酸杂质的序列覆盖率可达100%,根据UV峰面积测量,可测序的丰度水平低至0.2%。
- 使用适当的碰撞能量梯度进行数据非依赖型采集(MSE)可对FLP提供100%序列覆盖率,而无需对单个寡核苷酸母离子进行碰撞能量优化。
- CONFIRM Sequence应用程序能够查找源自FLP序列的序列省略、插入或序列变体。
参考资料
- Sharma VK, Watts JK Oligonucleotide therapeutics: chemistry, delivery and clinical progress, Future Med Chem, 2015, 7(16), 2221-2242.
- Roberts TK, Langer R, Wood MJA Advances in oligonucleotide drug delivery, Nat Reviews, 2020, 19, 673-694.
- Pourshahian S Therapeutic oligonucleotides, impurities, degradants, and their characterization by mass spectrometry, Mass Spectrometry Reviews, 2019, 00, 1–35.
- Obika S, Sekine M – editors of Synthesis of therapeutic oligonucleotides, 1st edition, Springer, 2018.
- Capaldi D, Teasdale A, Henry S, Akhtar N, den Besten C, Gao-Sheridan S, Kretschmer M, Sharpe N, Andrews B, Burm B, Foy J Impurities in oligonucleotide drug substances and drug products, impurities, degradants, and their characterization by mass spectrometry, Nucleic Acid Ther, 2017, 27, 309–322.
- 利用符合法规要求的自动化LC-MS工作流程对寡核苷酸进行完整质量数确认和纯度分析, 沃特世应用纪要, 720006820ZH, 2020.
- 使⽤BioAccord LC-MS系统对各种经过⼤量修饰的寡核苷酸进⾏完整质量数确认分析, 沃特世应用纪要, 720007028ZH, 2020.
- 使用装配ACQUITY Premier产品的BioAccord LC-MS系统分析寡核苷酸杂质, 沃特世应用纪要, 720007301ZH, 2021.
- 使用搭载ACQUITY Premier的BioAccord系统和全新的自动化INTACT Mass应用程序对siRNA、单向导RNA和杂质进行LC-MS分析, 沃特世应用纪要, 720007546ZH, 2022.
- McLukey SA, Van Berkel GJ, Glish GL Tandem Mass Spectrometry of Small, Multiply Charged Oligonucleotides, J Am Soc Mass Spectrom, 1992, 3, 60–70.
- Rozenski J, McCloskey J SOS: A simple interactive program for ab initio oligonucleotide sequencing by mass spectrometry, J Am Soc Mass Spectrom, 2002, 13, 200–203.
- Oberacher H, Parson W, Oefner PJ, Mayr BM, Huber CC Applicability of tandem mass spectrometry to the automated comparative sequencing of long-chain oligonucleotides, J Am Soc Mass Spectrom, 2004, 15, 510–522.
- Kretschmer M, Lavine G, McArdle J, Kuchimanchi S, Murugaiah V, Manoharan M An automated algorithm for sequence confirmation of chemically modified oligonucleotides by tandem mass spectrometry, Anal Biochem, 2010, 405, 213–223.
- Nakayama H, Akiyama M, Taoka M, Yamauchi Y, Nobe Y, Ishikawa H, Takahashi N, Isobe T Ariadne: a database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data Nucleic Acids Res, 2009, 37, 1–13.
- Yang J, Leopold P, Helmy R, Parish C, Arvary B, Mao B, Meng F Design and application of an easy to use oligonucleotide mass calculation program, J Am Soc Mass Spectrom, 2013, 24, 1315–1318.
- Nyakas A, Blum LC, Stucki SR, Reymond JL, Schurch S OMA and OPA – software -supported mass spectra analysis of native and modified nucleic acids, J Am Soc Mass Spectrom, 2013, 24, 249–256.
- Rozenski J Mongo oligo mass calculator.网址:http://rna.rega.kuleuven.be/masspec/mongo.htm.2022年6月22日访问.
- Kass, I Spectrum Tools, 2018.
- Rentel C, DaCosta J, Roussis S, Chan J, Capaldi D, Mai B Determination of oligonucleotide deamination by high resolution mass spectrometry, J Pharm Biomed Anal, 2019, 173, 56-61.
720007677ZH,2022年7月